Predicción del destino de las células
Los investigadores desarrollan soluciones de IA para la investigación médica de nueva generación
Los datos no sólo son la respuesta a numerosas preguntas en el mundo empresarial; lo mismo ocurre con la investigación biomédica. Para desarrollar nuevas terapias o estrategias de prevención de enfermedades, los científicos necesitan más y mejores datos, cada vez más rápido. Sin embargo, la calidad suele ser muy variable y la integración de diferentes conjuntos de datos, casi imposible. Con el Centro de Salud Computacional del Helmholtz de Múnich se está creando uno de los mayores centros de investigación de Europa sobre inteligencia artificial en ciencias médicas, bajo la dirección de Fabian Theis. En estrecha colaboración con la Universidad Técnica de Múnich (TUM), más de un centenar de científicos utilizan la inteligencia artificial y el aprendizaje automático para descubrir soluciones precisamente a estos problemas, permitiendo así innovaciones médicas para una sociedad más sana. En el último número de la revista Nature Methods, presentan tres artículos con nuevas e innovadoras soluciones.
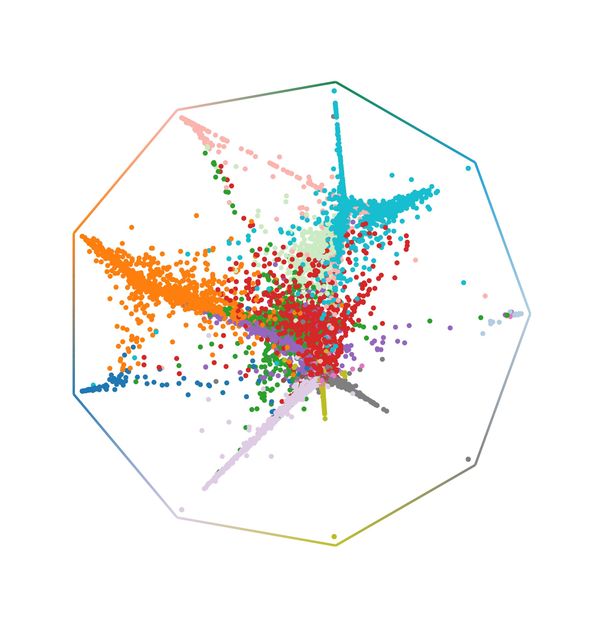
Probabilidades de destino de CellRank para la regeneración pulmonar; cada célula se coloca en una posición que refleja su probabilidad de alcanzar cualquier estado terminal.
© Helmholtz Munich / Marius Lange
Fabian Theis, director del Centro de Salud Computacional del Helmholtz de Múnich y catedrático de Modelización Matemática de Sistemas Biológicos de la TUM: "Han sido cuatro semanas de locura, en las que se han materializado muchos de nuestros métodos e historias científicas. Nuestros grupos de investigación se centran en el uso de la genómica unicelular para comprender el origen de las enfermedades de forma mecanicista, para lo cual aprovechamos y desarrollamos enfoques de aprendizaje automático para representar mejor estos datos complejos. En los tres nuevos artículos, trabajamos en la integración de datos de células individuales, el aprendizaje de trayectorias y la resolución espacial, respectivamente. Además de las aplicaciones mostradas en los artículos, esperamos apoyar la próxima generación de investigación de células individuales hacia la comprensión de enfermedades".
Estas son las últimas soluciones desarrolladas por los investigadores de Helmholtz Múnich y la TUM:
Resolver el reto de la integración de datos
Para ver si una observación que se hace en un solo conjunto de datos puede generalizarse, se puede comprobar si lo mismo puede observarse en otros conjuntos de datos del mismo sistema. En los datos unicelulares, los llamados efectos de lote complican la combinación de conjuntos de datos de esta manera. Se trata de diferencias en los perfiles moleculares entre las muestras, ya que se generaron en un momento diferente, en un lugar diferente o de una persona diferente. Superar estos efectos es un reto central en la genómica unicelular con más de 50 soluciones propuestas. Pero, ¿cuál es la mejor? Un grupo de investigadores en torno a Malte Lücken ha elaborado cuidadosamente 86 conjuntos de datos y ha comparado 16 de los métodos de integración de datos más populares en 13 tareas. Tras más de 55.000 horas de cálculo y una evaluación detallada de 590 resultados, construyeron una guía para la integración optimizada de datos. Esto permite mejorar las observaciones sobre los procesos de las enfermedades en los conjuntos de datos a escala poblacional.
Predicción de estados celulares con software de código abierto
Muchas cuestiones de la biología giran en torno a procesos continuos como el desarrollo o la regeneración. La secuenciación de ARN unicelular mide la expresión génica de cualquier célula que se encuentre en un proceso de este tipo. Sin embargo, este método es destructivo para las células y los científicos sólo obtienen instantáneas estáticas. Por ello, se han desarrollado muchos algoritmos para reconstruir procesos continuos a partir de instantáneas de la expresión génica. Una limitación común: Estos algoritmos no pueden decirnos nada sobre la dirección del proceso. Para superar esta limitación, Marius Lange y sus colegas desarrollaron un nuevo algoritmo llamado CellRank. Este algoritmo estima las trayectorias dirigidas del estado celular combinando los enfoques de reconstrucción anteriores con la velocidad del ARN, un concepto para estimar la regulación ascendente o descendente de los genes. En las aplicaciones in vitro e in vivo, CellRank infirió correctamente los resultados del destino y recuperó genes previamente conocidos. En un ejemplo de regeneración pulmonar, CellRank predijo nuevos estados celulares intermedios en una trayectoria de desdiferenciación cuya existencia fue validada experimentalmente. CellRank es un paquete de software de código abierto que ya utilizan biólogos y bioinformáticos de todo el mundo para analizar dinámicas celulares complejas en situaciones como el cáncer, la reprogramación o la regeneración.
Visualización del análisis ómico espacial
En los últimos años se han desarrollado cada vez más tecnologías para medir la variación de la expresión génica en los tejidos. La ventaja de estas tecnologías es que los científicos pueden ver las células en su contexto, pudiendo así investigar los principios de la organización de los tejidos y la comunicación celular. Los investigadores necesitan marcos computacionales flexibles para almacenar, integrar y visualizar la creciente diversidad de estos datos. Para hacer frente a este reto, Giovanni Palla, Hannah Spitzer y sus colegas han desarrollado un nuevo marco computacional, llamado Squidpy. Permite a los analistas y desarrolladores manejar datos espaciales de expresión génica. Squidpy integra herramientas de expresión génica y de análisis de imágenes para manipular eficazmente y visualizar de forma interactiva los datos ómicos espaciales. Squidpy es extensible y puede interconectarse con una variedad de herramientas de aprendizaje automático en el ecosistema de python. Científicos de todo el mundo ya lo utilizan para analizar datos moleculares espaciales.
Nota: Este artículo ha sido traducido utilizando un sistema informático sin intervención humana. LUMITOS ofrece estas traducciones automáticas para presentar una gama más amplia de noticias de actualidad. Como este artículo ha sido traducido con traducción automática, es posible que contenga errores de vocabulario, sintaxis o gramática. El artículo original en Inglés se puede encontrar aquí.
Publicación original
Más noticias del departamento ciencias




















Estos productos pueden interesarle

Limsophy de AAC Infotray
Optimice los procesos de su laboratorio con Limsophy LIMS
Integración perfecta y optimización de procesos en la gestión de datos de laboratorio
ERP-Software de GUS
GUS-OS Suite: Su ERP para la industria de procesos
Optimizamos sus procesos empresariales































