Desbloquear los secretos de los genomas de las plantas en alta resolución
La nueva herramienta de software permite una asignación precisa a las copias correctas, un proceso conocido como "phasing".
Resolver los genomas, en particular los de las plantas, es una tarea muy compleja y propensa a errores. Esto se debe a que hay varias copias de todos los cromosomas y son muy similares. Un equipo de investigadores en bioinformática de la Universidad Heinrich Heine de Düsseldorf (HHU) ha desarrollado una herramienta de software que permite una asignación precisa a las copias correctas, un proceso conocido como "phasing".

La nueva herramienta de software permite determinar el genoma de especies como las patatas con un alto grado de precisión (imagen simbólica).
Capri23auto, pixabay.com

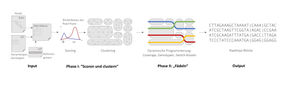
Cómo funciona la herramienta de software bioinformático 'WhatsHap polyphase'.
Gunnar Klau/HHU

Los genomas de todas las formas de vida superiores se almacenan en el núcleo de la célula en los cromosomas. Los cromosomas están compuestos por cadenas de la molécula de ADN. La información genética propiamente dicha está codificada en una secuencia de pares de bases adyacentes de las moléculas adenina (A), citosina (C), guanina (G) y timina (T).
Las diferentes especies tienen diferentes números de cromosomas; por ejemplo, los humanos tienen 23, mientras que las patatas tienen 12 y el trigo 7. Además, hay diferentes copias o "haplotipos" de los cromosomas. Los humanos tienen dos copias, una de la madre y otra del padre, mientras que las patatas tienen cuatro y el trigo incluso seis. Las especies con dos copias se denominan "diploides", mientras que las que tienen más de dos son "poliploides". Las copias son casi idénticas, siendo "casi" la palabra operativa. Son las diferencias entre ellas las que determinan la variabilidad de los organismos dentro de una población.

Con el fin de desbloquear la información genética, los investigadores abordaron algo parecido a un gran rompecabezas: Tomaron un mayor número de células, dividieron los genomas de las células en muchas partes pequeñas - llamadas "lecturas" - y secuenciaron la información contenida en esas partes. Esto fue necesario porque la tecnología disponible actualmente sólo puede procesar pequeñas secciones de ADN.
El resultado fue un enorme volumen de datos - miles de millones de lecturas, con un volumen de datos de varios cientos de gigabytes. Comprenden secuencias de diferentes longitudes compuestas por las letras A, C, G y T. La siguiente tarea de los investigadores en bioinformática fue determinar su posición dentro de un cromosoma, luego asignar las secciones correspondientes a un cromosoma (un proceso conocido como "mapeo") y finalmente encontrar las copias correctas del cromosoma. Esta última etapa se conoce como 'phasing'. La tarea se hace más difícil debido a los errores de secuenciación.
Hay herramientas buenas y eficientes disponibles para la cartografía. Sin embargo, las herramientas bioinformáticas necesarias para el escalonamiento están todavía en su infancia. Aquí fue precisamente donde el equipo de investigadores bioinformáticos de HHU centró su atención. En un proyecto conjunto financiado por la Fundación Alemana de Investigación y dirigido por el Prof. Dr. Gunnar Klau (grupo de trabajo de Bioinformática Algorítmica) y el Prof. Dr. Tobias Marschall (Instituto de Biometría Médica y Bioinformática, Hospital Universitario de Düsseldorf) en colaboración con el Prof. Dr. Björn Usadel (Instituto de Ciencia de Datos Biológicos), desarrollaron una herramienta de software llamada 'WhatsHap polyphase' y probaron la herramienta con éxito utilizando datos de modelo así como el genoma de la patata.
Esta nueva herramienta resuelve el problema utilizando un proceso de dos fases. La primera fase consiste en agrupar las lecturas, es decir, dividirlas en grupos. Las lecturas en un grupo probablemente provengan de un haplotipo o de una región de haplotipos idénticos. La segunda fase implica "enhebrar" los haplotipos a través de los grupos. Durante el enhebrado, las lecturas se asignan a los haplotipos de la forma más uniforme posible, asegurando que se salte lo menos posible entre los grupos.
La nueva herramienta se ha añadido al paquete principal de "WhatsHap", que está disponible gratuitamente. El paquete ya se ha utilizado para llevar a cabo la fase con éxito para conjuntos de cromosomas diploides, por ejemplo, para los seres humanos. Esta nueva adición del equipo con sede en Düsseldorf significa que el phasing es ahora también posible para los organismos poliploides. El Prof. Klau dijo: "Nuestra nueva tecnología permite que los genomas de las plantas sean introducidos en fase de alta resolución y con un bajo margen de error".
Nota: Este artículo ha sido traducido utilizando un sistema informático sin intervención humana. LUMITOS ofrece estas traducciones automáticas para presentar una gama más amplia de noticias de actualidad. Como este artículo ha sido traducido con traducción automática, es posible que contenga errores de vocabulario, sintaxis o gramática. El artículo original en Inglés se puede encontrar aquí.
Publicación original

Más noticias del departamento ciencias



















Estos productos pueden interesarle

Limsophy de AAC Infotray
Optimice los procesos de su laboratorio con Limsophy LIMS
Integración perfecta y optimización de procesos en la gestión de datos de laboratorio
ERP-Software de GUS
GUS-OS Suite: Su ERP para la industria de procesos
Optimizamos sus procesos empresariales































