Comprendre comment les mutations affectent les maladies
Un nouveau cadre pour étudier les maladies polygéniques
Annonces



Le code génétique humain est entièrement cartographié, ce qui permet aux scientifiques de disposer d'un plan de l'ADN pour identifier les régions génomiques et leurs variations responsables de maladies. Les outils statistiques traditionnels permettent de repérer efficacement ces "aiguilles dans la botte de foin" génétique, mais ils se heurtent à des difficultés lorsqu'il s'agit de comprendre le nombre de gènes qui contribuent aux maladies, comme dans le cas du diabète ou de la schizophrénie.
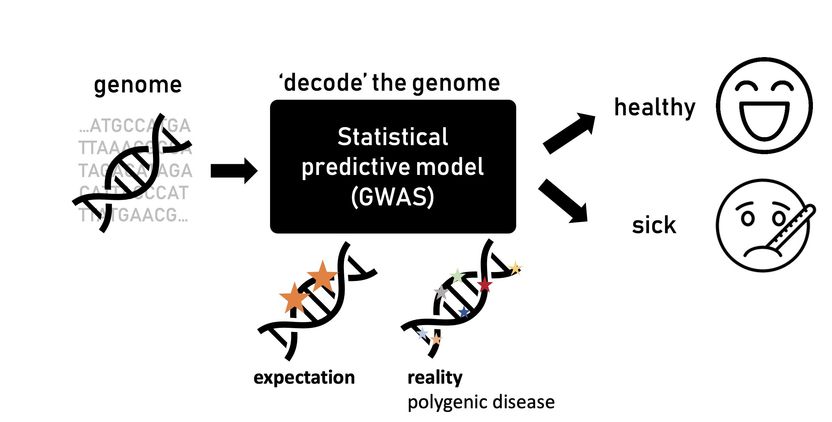
Les modèles traditionnels de l'étude d'association pangénomique (GWAS), qui s'apparentent à une "boîte noire", utilisent des statistiques pour prédire le risque de maladie à partir des génomes. La plupart des maladies sont polygéniques, c'est-à-dire que de nombreux sites du génome contribuent au risque de maladie, ce qui rend difficile l'identification des mécanismes biologiques sous-jacents.
Natália Ružičková / ISTA
De nombreux modèles statistiques et algorithmes utilisés par les scientifiques peuvent être comparés à une "boîte noire". Ces modèles sont des outils puissants qui donnent des prédictions précises, mais leur fonctionnement interne n'est pas facile à interpréter ou à comprendre. À une époque dominée par le deep learning, où une quantité toujours plus importante de données peut être traitée, Natália Ružičková, physicienne et doctorante à l'Institut des sciences et technologies d'Autriche (ISTA), a choisi de prendre du recul. Du moins dans le contexte de l'analyse des données génomiques.
Avec Michal Hledík, récemment diplômé de l'ISTA, et le professeur Gašper Tkačik, Ružičková a maintenant proposé un modèle qui pourrait aider à analyser les "maladies polygéniques", où de nombreuses régions du génome contribuent à un dysfonctionnement. Le modèle permet également de comprendre pourquoi les régions génomiques identifiées contribuent à ces maladies. Ils y parviennent en combinant une analyse du génome à la pointe de la technologie et des connaissances en biologie fondamentale. Les résultats sont publiés dans PNAS.
Décoder le génome humain
En 1990, le projet du génome humain a été lancé pour décoder entièrement l'ADN humain, le plan génétique qui définit l'homme. En 2003, lorsque le projet a été achevé, il a ouvert la voie à de nombreuses percées dans les domaines de la science, de la médecine et de la technologie. En déchiffrant le code génétique humain, les scientifiques espéraient en apprendre davantage sur les maladies liées à des mutations et à des variations spécifiques de ce script génétique. Étant donné que le génome humain comprend environ 20 000 gènes et encore plus de paires de bases - les lettres de l'empreinte bleue -, il était essentiel de disposer d'une grande puissance statistique. C'est ainsi qu'ont été développées les "études d'association à l'échelle du génome" (GWAS).
Les études d'association pangénomique abordent la question en identifiant les variantes génétiques potentiellement liées à des caractéristiques de l'organisme telles que la taille. Il est important de noter qu'elles incluent également la propension à diverses maladies. Pour ce faire, le principe statistique sous-jacent est assez simple : les participants sont divisés en deux groupes, les individus sains et les individus malades. Leur ADN est ensuite analysé afin de détecter les variations - changements dans leur génome - qui sont plus marquées chez les personnes atteintes de la maladie.
Une interaction de gènes
Lorsque les études d'association à l'échelle du génome sont apparues, les scientifiques s'attendaient à ne trouver que quelques mutations dans des gènes connus liés à une maladie, qui expliqueraient la différence entre les individus sains et les individus malades. En réalité, la situation est beaucoup plus complexe. "Parfois, il y a des centaines ou des milliers de mutations liées à une maladie spécifique", explique Ružičková. "C'était une révélation surprenante et contradictoire avec la compréhension de la biologie que nous avions".
Individuellement, chaque mutation a un impact ou une contribution minime sur le risque de développer une maladie. Cependant, collectivement, elles peuvent mieux expliquer, mais pas entièrement, pourquoi certains individus développent la maladie. Ces maladies sont dites "polygéniques". Par exemple, le diabète de type 2 est polygénique, car il ne peut être attribué à un seul gène ; il implique au contraire des centaines de mutations. Certaines de ces mutations affectent la production d'insuline, l'action de l'insuline ou le métabolisme du glucose, tandis que la majorité d'entre elles sont situées dans des régions génomiques qui n'ont jamais été liées au diabète ou dont les fonctions biologiques sont inconnues.
Le modèle omnigène
En 2017, Evan A. Boyle et ses collègues de l'université de Stanford ont proposé un nouveau cadre conceptuel appelé "modèle omnigénique". Ils ont proposé une explication à la raison pour laquelle tant de gènes contribuent aux maladies : les cellules possèdent des réseaux de régulation qui relient des gènes aux fonctions diverses.
"Comme les gènes sont interconnectés, une mutation dans un gène peut avoir un impact sur d'autres gènes, car l'effet de la mutation se propage dans le réseau de régulation", explique Ružičková. Grâce à ces réseaux, de nombreux gènes du système de régulation finissent par contribuer à une maladie. Cependant, jusqu'à présent, ce modèle n'a pas été formulé mathématiquement et est resté une hypothèse conceptuelle difficile à tester. Dans leur dernier article, Ružičková et ses collègues présentent une nouvelle formalisation mathématique basée sur le modèle omnigène, appelée "modèle omnigène quantitatif" (QOM).
Combiner statistiques et biologie
Pour démontrer le potentiel du nouveau modèle, ils ont dû appliquer le cadre à un système biologique bien caractérisé. Ils ont choisi le modèle de levure de laboratoire commun Saccharomyces cerevisiae, mieux connu sous le nom de levure de bière ou de levure de boulanger. Il s'agit d'un eucaryote unicellulaire, ce qui signifie que sa structure cellulaire est similaire à celle d'organismes complexes tels que l'homme. "Dans la levure, nous comprenons assez bien comment sont structurés les réseaux de régulation qui interconnectent les gènes", explique Ružičková.
À l'aide de leur modèle, les scientifiques ont prédit les niveaux d'expression des gènes - l'intensité de l'activité des gènes, qui indique la quantité d'informations de l'ADN activement utilisée - et la façon dont les mutations se propagent dans le réseau de régulation de la levure. Les prédictions ont été très efficaces : Le modèle a non seulement identifié les gènes pertinents, mais il a également pu déterminer avec précision quelle mutation avait le plus de chances de contribuer à un résultat spécifique.
Les pièces du puzzle des maladies polygéniques
L'objectif des scientifiques n'était pas de surpasser le GWAS standard en termes de performances de prédiction, mais plutôt d'aller dans une direction différente en rendant le modèle interprétable. Alors qu'un modèle GWAS standard fonctionne comme une "boîte noire", offrant un compte rendu statistique de la fréquence à laquelle une mutation particulière est liée à une maladie, le nouveau modèle fournit également un mécanisme causal de la chaîne d'événements qui explique comment cette mutation peut conduire à une maladie.
En médecine, la compréhension du contexte biologique et de ces voies de causalité a d'énormes implications pour la recherche de nouvelles options thérapeutiques. Bien que le modèle soit actuellement loin de toute application médicale, il présente un potentiel, notamment pour en savoir plus sur les maladies polygéniques. "Si l'on dispose de suffisamment de connaissances sur les réseaux de régulation, on peut construire des modèles similaires pour d'autres organismes. Nous avons étudié l'expression des gènes dans la levure, ce qui n'est qu'une première étape et une preuve de principe. Maintenant que nous comprenons ce qui est possible, on peut commencer à penser aux applications à la génétique humaine", explique Ružičková.
Note: Cet article a été traduit à l'aide d'un système informatique sans intervention humaine. LUMITOS propose ces traductions automatiques pour présenter un plus large éventail d'actualités. Comme cet article a été traduit avec traduction automatique, il est possible qu'il contienne des erreurs de vocabulaire, de syntaxe ou de grammaire. L'article original dans Anglais peut être trouvé ici.
Publication originale

Annonces



Autres actualités du département science













































