Des milliers d'heures à quelques minutes : un logiciel accélère le développement des médicaments
Des chercheurs développent une nouvelle méthode pour prédire en quelques minutes la morphologie des couches de sucre sur des protéines cliniquement pertinentes

Les sucres recouvrent presque toutes les protéines présentes à la surface des cellules de notre corps, formant un bouclier autour des protéines. Ces sucres influencent donc la manière dont les cellules interagissent avec leur environnement, y compris les agents pathogènes, et jouent un rôle important dans le développement de médicaments. GlycoSHIELD, une nouvelle approche computationnelle pour étudier les boucliers de sucre des protéines, permet de réduire les ressources, de gagner du temps et d'être convivial. Elle a été développée dans le cadre du programme Dioscuri de la société Max Planck dans un projet de coopération germano-polonais, grâce à des collaborations internationales avec des chercheurs en France, à Taiwan et en Allemagne.
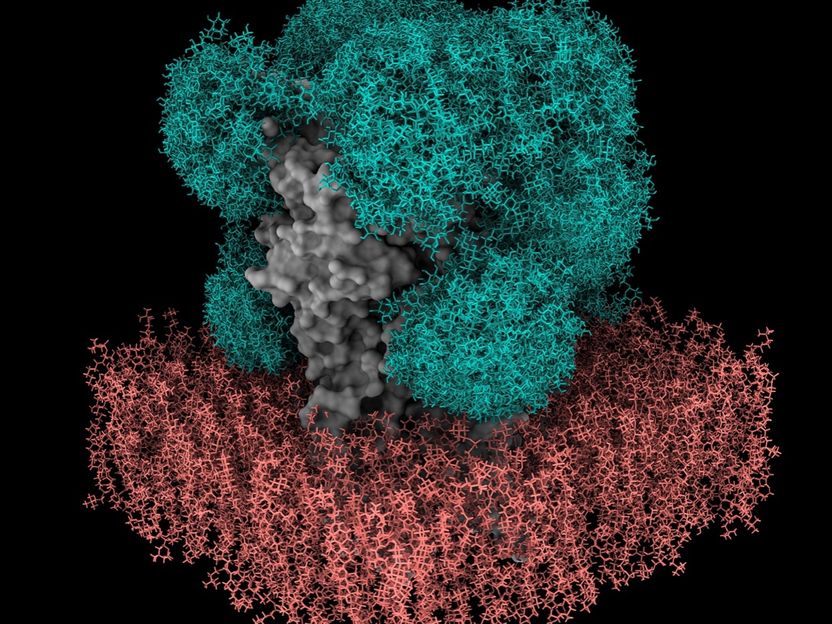
Modèle du bouclier de sucre (vert) sur le récepteur GABAA (gris) dans une membrane (rouge) généré par GlycoSHIELD.
© Dr. Cyril Hanus, Inserm, University Paris-Cité.
Les protéines remplissent non seulement des fonctions essentielles à la survie des cellules, mais elles influencent également le développement et la progression des maladies. Pour comprendre leur rôle dans la santé et la maladie, les chercheurs étudient la structure atomique tridimensionnelle des protéines à l'aide de méthodes expérimentales et informatiques. Plus de 75 % des protéines présentes à la surface de nos cellules sont recouvertes de glycanes. Ces molécules ressemblant à des sucres forment des boucliers protecteurs très dynamiques autour des protéines. Cependant, la mobilité et la variabilité des sucres rendent difficile la détermination du comportement de ces boucliers ou de leur influence sur la fixation des molécules médicamenteuses. Mateusz Sikora, chef de projet et directeur du Centre Dioscuri pour la modélisation des modifications post-traductionnelles, et son équipe de Cracovie ainsi que ses partenaires de l'Institut Max Planck de biophysique de Francfort-sur-le-Main, en Allemagne, ont relevé ce défi en utilisant des ordinateurs, en collaboration avec des scientifiques de l'Inserm à Paris, de l'Academia Sinica à Tapei et de l'Université de Brême. Leur nouvel algorithme puissant, GlycoSHIELD, permet une modélisation rapide et réaliste des chaînes de sucres présentes sur les surfaces des protéines. Réduisant les heures de calcul et donc la consommation d'énergie de plusieurs ordres de grandeur par rapport aux outils de simulation conventionnels, GlycoSHIELD ouvre la voie à l'informatique verte. Les scientifiques ont publié leur nouvelle méthode et sa validation dans la revue Cell.
Des milliers d'heures à quelques minutes
Les glycanes protecteurs influencent fortement la façon dont les protéines interagissent avec d'autres molécules telles que les médicaments thérapeutiques. Par exemple, la couche de sucre sur la protéine spike du coronavirus cache le virus au système immunitaire en rendant difficile la reconnaissance du virus par les anticorps naturels ou induits par la vaccination. Les boucliers de sucre jouent donc un rôle important dans le développement de médicaments et de vaccins. La recherche pharmaceutique pourrait bénéficier d'une prévision systématique de leur morphologie et de leur dynamique. Jusqu'à présent, la prévision de la structure des couches de sucre à l'aide de simulations informatiques n'était possible qu'avec des connaissances spécialisées sur des superordinateurs spéciaux. Dans de nombreux cas, des milliers, voire des millions d'heures de calcul étaient nécessaires. Avec GlycoSHIELD, l'équipe de M. Sikora propose une alternative open source rapide et respectueuse de l'environnement. "Notre approche réduit les ressources, le temps de calcul et l'expertise technique nécessaire", explique M. Sikora. "N'importe qui peut désormais calculer l'arrangement et la dynamique des molécules de sucre sur les protéines sur son ordinateur personnel en quelques minutes, sans avoir besoin de connaissances spécialisées ni d'ordinateurs très performants. En outre, cette nouvelle méthode de calcul est très économe en énergie". Le logiciel peut non seulement être utilisé dans la recherche, mais il pourrait également être utile pour le développement de médicaments ou de vaccins, par exemple dans le cadre de l'immunothérapie contre le cancer.
Un puzzle en sucre
Comment l'équipe a-t-elle réussi à obtenir une telle augmentation de l'efficacité ? Les auteurs ont créé et analysé une bibliothèque de milliers de poses 3D les plus probables des formes les plus courantes de chaînes de sucre sur des protéines présentes chez l'homme et les micro-organismes. À l'aide de longues simulations et expériences, ils ont constaté que pour une prédiction fiable des boucliers glycanniques, il suffit que les sucres attachés n'entrent pas en collision avec des membranes ou des parties de la protéine. L'algorithme est basé sur ces résultats. "Les utilisateurs de GlyoSHIELD n'ont qu'à spécifier la protéine et les endroits où les sucres sont attachés. Notre logiciel les place ensuite à la surface de la protéine selon la disposition la plus probable", explique M. Sikora. "Nous avons pu reproduire avec précision les boucliers de sucres de la protéine spike : ils ressemblent exactement à ce que nous voyons dans les expériences ! Avec GlycoSHIELD, il est désormais possible d'ajouter des informations sur les sucres aux structures protéiques nouvelles ou existantes. Les scientifiques ont également utilisé GlycoSHIELD pour révéler le schéma des sucres sur le récepteur GABAA, une cible importante pour les sédatifs et les anesthésiques.
Un succès pour le Centre Dioscuri
Les centres Dioscuri initiés par la Société Max Planck ont pour but de renforcer et d'étendre l'excellence de la recherche en Europe centrale et orientale. Depuis mai 2023, Mateusz Sikora, ancien chercheur postdoctoral à l'Institut Max Planck de biophysique, bénéficie d'un soutien financier dans le cadre du programme financé bilatéralement en tant que responsable du Centre Dioscuri pour la modélisation des modifications post-traductionnelles créé à l'Université Jagiellonian de Cracovie, en Pologne. Gerhard Hummer, directeur du département de biophysique théorique à l'Institut Max Planck de biophysique, le soutient en tant que partenaire depuis l'Allemagne et a également contribué à ce travail. Après moins d'un an, M. Sikora a déjà remporté un grand succès avec son algorithme vert et contribue à promouvoir la Pologne en tant que site de recherche attrayant et compétitif.
Note: Cet article a été traduit à l'aide d'un système informatique sans intervention humaine. LUMITOS propose ces traductions automatiques pour présenter un plus large éventail d'actualités. Comme cet article a été traduit avec traduction automatique, il est possible qu'il contienne des erreurs de vocabulaire, de syntaxe ou de grammaire. L'article original dans Anglais peut être trouvé ici.
Publication originale
Yu-Xi Tsai, Ning-En Chang, Klaus Reuter, Hao-Ting Chang, Tzu-Jing Yang, Sören von Bülow, Vidhi Sehrawat, Noémie Zerrouki, Matthieu Tuffery, Michael Gecht, Isabell Louise Grothaus, Lucio Colombi Ciacchi, Yong-Sheng Wang, Min-Feng Hsu, Kay-Hooi Khoo, Gerhard Hummer, Shang-Te Danny Hsu, Cyril Hanus, Mateusz Sikora; "Rapid simulation of glycoprotein structures by grafting and steric exclusion of glycan conformer libraries"; Cell, Volume 187

Autres actualités du département science




















Ces produits pourraient vous intéresser

Limsophy de AAC Infotray
Optimisez vos processus de laboratoire avec Limsophy LIMS
Intégration transparente et optimisation des processus dans la gestion des données de laboratoire
ERP-Software de GUS
GUS-OS Suite : votre ERP pour l'industrie de process
Nous optimisons les processus de votre entreprise