Un nouvel outil facilite l'interprétation clinique des informations génétiques
Annonces


Les équipes de recherche de Max Planck et de Harvard développent DeMAG, une nouvelle méthode partagée sous la forme d'un serveur web open-source (demag.org) pour aider à interpréter les mutations dans les gènes pathologiques et améliorer la prise de décision clinique.
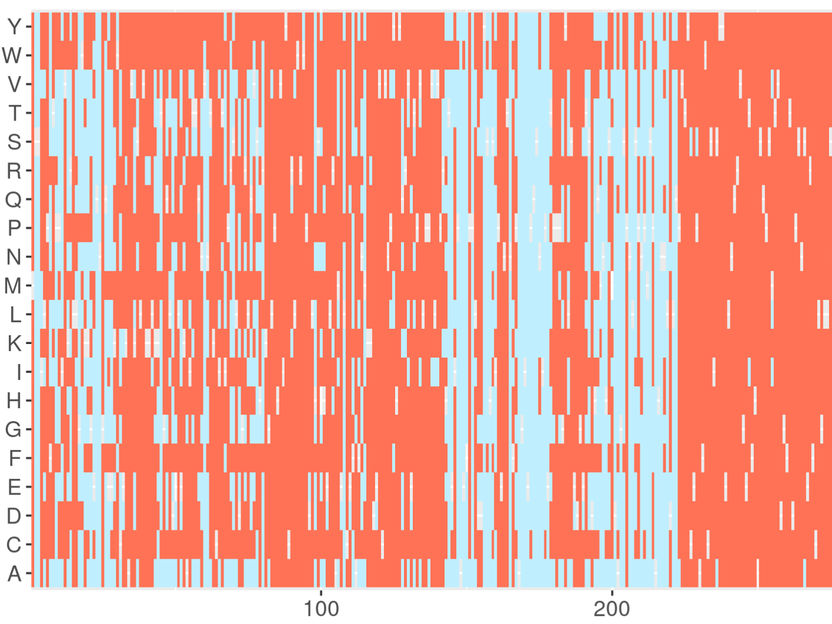
Capture d'écran du serveur web DeMAG. DeMAG prédit les mutations bénignes en bleu clair et les mutations pathogènes en corail.
Agnes Toth-Petroczy, Nature Communications, 2023 / MPI-CBG
Malgré l'utilisation croissante du séquençage génomique dans la pratique clinique, l'interprétation des mutations génétiques rares, même parmi les gènes pathologiques bien étudiés, reste difficile. Les modèles prédictifs actuels sont utiles pour interpréter ces mutations, mais ils ont tendance à mal classer celles qui ne causent pas de maladies, ce qui contribue à des faux positifs. Des chercheurs de l'Institut Max Planck de biologie cellulaire moléculaire et de génétique (MPI-CBG) de Dresde, du Centre de biologie des systèmes de Dresde (CSBD) en Allemagne et de la Harvard Medical School de Boston, aux États-Unis, ont mis au point un outil appelé Deciphering Mutations in Actionable Genes (DeMAG), publié dans la revue Nature Communications. DeMAG est un serveur web à code source ouvert qui offre une interprétation des effets de toutes les mutations potentielles d'un seul acide aminé pouvant survenir dans 316 gènes cliniquement pertinents qui causent des maladies pour lesquelles des diagnostics et des traitements préventifs sont déjà disponibles. DeMAG fournit aux professionnels de la santé un outil qui leur permet d'évaluer plus précisément l'effet des mutations dans ces gènes en réduisant le taux de faux positifs, ce qui signifie que des mutations moins bénignes sont prédites comme pathogènes. Cet outil peut donc faciliter la prise de décision clinique.
Ces dernières années, le séquençage génomique est devenu moins coûteux et plus avancé. D'une part, cela permet aux cliniciens d'utiliser de plus en plus le séquençage à des fins de diagnostic tout en permettant aux scientifiques d'explorer davantage d'hypothèses de recherche. D'autre part, de nombreuses mutations détectées n'ont pas d'interprétation clinique claire. L'incertitude quant à savoir si une mutation est à l'origine d'une maladie peut être stressante pour les patients et entraîner une charge psychologique, une morbidité et des dépenses de santé associées à un sous-diagnostic ou à un surdiagnostic. Bien que les outils existants soient déjà utilisés pour prédire l'impact fonctionnel de ces variantes, leurs performances sont biaisées en raison du manque de données cliniques qui rend difficile la distinction entre les variantes pathogènes (causant la maladie) et bénignes (neutres) au sein d'un gène donné et conduit souvent à classer à tort comme pathogènes des mutations qui ne causent pas de maladie. Il est essentiel de résoudre ces difficultés si l'on veut mettre au point un prédicteur fiable pour les applications cliniques.
Le groupe de recherche d'Agnes Toth-Petroczy au MPI-CBG et au CSBD a fait équipe avec Christopher Cassa, professeur adjoint de médecine à la division de génétique du Brigham and Women's Hospital de la Harvard Medical School, et Ivan Adzhubei, chercheur associé au département d'informatique biomédicale de la Harvard Medical School, pour développer un modèle statistique et le serveur web DeMAG qui permet d'obtenir une grande précision dans l'interprétation des mutations génétiques dans les gènes pathogènes. Pour ce faire, les chercheurs ont soigneusement sélectionné des mutations pathogènes et bénignes connues pour l'entraînement du modèle. "Nous avons utilisé des bases de données cliniques et de diverses populations. Nous n'avons sélectionné que des mutations dont l'interprétation clinique fait l'objet d'un consensus entre de multiples auteurs, tels que des médecins et des laboratoires de génétique. Nous avons également inclus des données provenant d'ancêtres sous-représentés dans les bases de données démographiques actuelles, comme les Coréens ou les Japonais, afin de rendre le modèle encore plus représentatif et précis", explique Federica Luppino, premier auteur de l'article de recherche et doctorante dans le groupe Toth-Petroczy. DeMAG comprend une nouvelle fonctionnalité, le "score de partenaires", qui identifie les groupes d'acides aminés dans une protéine qui partagent le même effet clinique. Avec le score de partenaires, DeMAG tire parti des relations entre les acides aminés basées sur les informations évolutives des génomes de nombreux organismes et de la récente révolution de l'IA (intelligence artificielle) consistant à prédire les formes 3D des protéines à l'aide de l'algorithme AlphaFold mis au point par Google DeepMind.
Agnes Toth-Petroczy, qui a supervisé l'étude, conclut : "Nous fournissons un cadre de base pour l'intégration des données cliniques et protéiques afin de faciliter l'évaluation de l'impact des mutations. Nous espérons que notre outil et notre serveur web faciliteront l'évaluation des effets des variantes et la prise de décision clinique. En outre, les nouvelles fonctionnalités développées peuvent être appliquées à d'autres gènes et organismes que l'homme".
Note: Cet article a été traduit à l'aide d'un système informatique sans intervention humaine. LUMITOS propose ces traductions automatiques pour présenter un plus large éventail d'actualités. Comme cet article a été traduit avec traduction automatique, il est possible qu'il contienne des erreurs de vocabulaire, de syntaxe ou de grammaire. L'article original dans Anglais peut être trouvé ici.
Publication originale




Autres actualités du département science






































