Un nouvel outil intègre l'analyse du microbiome et du séquençage génétique de l'hôte
Un nouveau logiciel facilite l'étude des relations entre un hôte, son microbiome et des agents pathogènes tels que le VIH ou le SRAS-CoV-2.
Annonces



Des chercheurs du Texas Biomedical Research Institute et de l'université de Tulane ont mis au point un nouvel outil logiciel qui rend plus facile, plus rapide et plus rentable l'analyse simultanée des informations génétiques d'un hôte et de son microbiome. Le logiciel, appelé "détecteur de méta-transcriptome" (MTD), peut être utilisé par un large éventail de microbiologistes et de développeurs de médicaments, notamment ceux qui étudient des maladies telles que certains cancers, le COVID-19, le VIH/SIDA, le paludisme et de nombreuses autres affections humaines liées aux micro-organismes. L'outil a récemment été publié dans la revue Briefings in Bioinformatics.
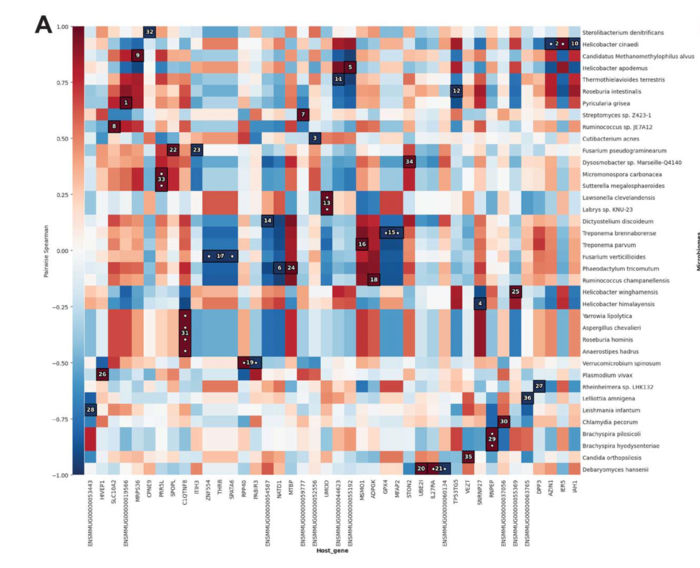
MTD analyse l'activité de l'expression génique dans l'hôte et le microbiome en même temps, et génère automatiquement un graphique montrant les corrélations entre les deux. Un rouge plus foncé indique une corrélation positive plus forte, ce qui signifie que la régulation positive ou négative d'un certain gène ou d'une certaine voie de l'hôte est liée à la régulation positive ou négative d'un certain type de microbe dans la même direction. Un bleu plus foncé indique une corrélation négative plus forte, ce qui signifie qu'un gène ou une voie de l'hôte est moins actif mais que l'espèce de microbe est plus active. Bien que l'analyse n'indique pas que l'un cause l'autre, elle peut mettre en évidence des relations que les chercheurs peuvent étudier plus avant et qui pourraient aider à comprendre la relation de cause à effet pour le développement potentiel de traitements.
Texas Biomed
"Il est très convivial, en particulier pour les chercheurs ayant peu ou pas de connaissances en bioinformatique", explique le professeur agrégé Binhua "Julie" Ling, qui codirige le programme de recherche sur les interactions hôte-pathogène de Texas Biomed et est l'auteur principal de l'article. "Il suffit d'écrire une ligne de code pour définir certains paramètres et le logiciel fait le reste automatiquement."
MTD permet aux chercheurs d'obtenir un instantané complet des microbes présents dans l'hôte, y compris la vaste gamme de "bonnes bactéries" qui vivent normalement sur et à l'intérieur des personnes et des animaux, ainsi que les bactéries nocives, comme les virus qui causent des maladies graves. Plus important encore, MTD analyse l'activité de l'expression génétique, c'est-à-dire les gènes qui sont activés ou désactivés, à la fois dans les microbes et dans l'hôte, ce qui permet aux chercheurs de repérer facilement les relations entre eux. Par exemple, voir quels gènes sont actifs dans un microbe et dans l'hôte pourrait indiquer que l'activité de l'un est influencée par l'autre, et pourrait suggérer une cible potentielle pour un traitement ultérieur, explique Ling.

"Nous ne pouvons pas dire à ce stade s'il s'agit d'une relation de cause à effet, mais nous pouvons utiliser cette analyse pour identifier les gènes ou les voies que nous devrions étudier - peut-être des gènes ou des voies que nous n'avions jamais envisagés auparavant comme étant liés", explique Ling. "MTD peut contribuer à accélérer ce processus et potentiellement ouvrir de nouvelles voies de recherche et de développement de médicaments."
MTD relie plusieurs logiciels existants et puise dans des bases de données internationales contenant des séquences d'ARN de plus de 100 000 espèces de bactéries, de virus, de champignons, d'archées et de protozoaires, ainsi que des séquences de vecteurs et de plasmides. Les utilisateurs peuvent également mettre à jour la base de données avec des séquences spécifiques qui les intéressent.
Le premier auteur de l'article, Fei Wu, a travaillé avec Ling pour étudier comment le microbiome évolue avec l'âge chez des singes jeunes et âgés atteints du virus de l'immunodéficience simienne (VIS), la version singe du VIH.
"Nous devions analyser l'expression génétique de l'hôte au moyen d'un flux de travail et l'expression génétique du microbiome au moyen d'un flux de travail distinct", explique M. Wu. "Nous nous sommes demandé pourquoi nous ne pouvions pas faire les deux en même temps".
Comme leur laboratoire était délocalisé pendant la pandémie de COVID-19, ils ont eu le temps de se concentrer sur le travail informatique, et ont entrepris de résoudre ce problème. Wu a travaillé avec Ling et son collaborateur Yao-Zhong Liu, PhD, professeur associé à l'école de santé publique et de médecine tropicale de l'université Tulane, pour construire et tester le nouveau logiciel.
"Normalement, nous utilisons des logiciels de bioinformatique pour analyser nos données, pas pour les construire", explique Wu. "C'était un défi, mais passionnant, de se diversifier et d'avoir maintenant quelque chose qui va non seulement nous aider, mais aussi tout autre chercheur faisant du séquençage d'ARN d'hôtes et de microbiomes ; des humains et des singes, aux moustiques porteurs de parasites du paludisme et aux escargots porteurs de parasites du schistosome."
Note: Cet article a été traduit à l'aide d'un système informatique sans intervention humaine. LUMITOS propose ces traductions automatiques pour présenter un plus large éventail d'actualités. Comme cet article a été traduit avec traduction automatique, il est possible qu'il contienne des erreurs de vocabulaire, de syntaxe ou de grammaire. L'article original dans Anglais peut être trouvé ici.
Publication originale



Autres actualités du département science



















Ces produits pourraient vous intéresser