Los investigadores estudian los cambios estructurales de las proteínas congeladas
Un novedoso método permite visualizar los movimientos moleculares que duran unas millonésimas de segundo
Investigadores de la Universidad de Bonn y del centro de investigación Caesar han conseguido congelar proteínas de forma ultrarrápida tras un periodo de tiempo definido con precisión. Pudieron seguir los cambios estructurales en la escala de tiempo de microsegundos y con una precisión subnanométrica. Gracias a su alta resolución espacial y temporal, el método permite seguir cambios estructurales rápidos en enzimas y ácidos nucleicos.
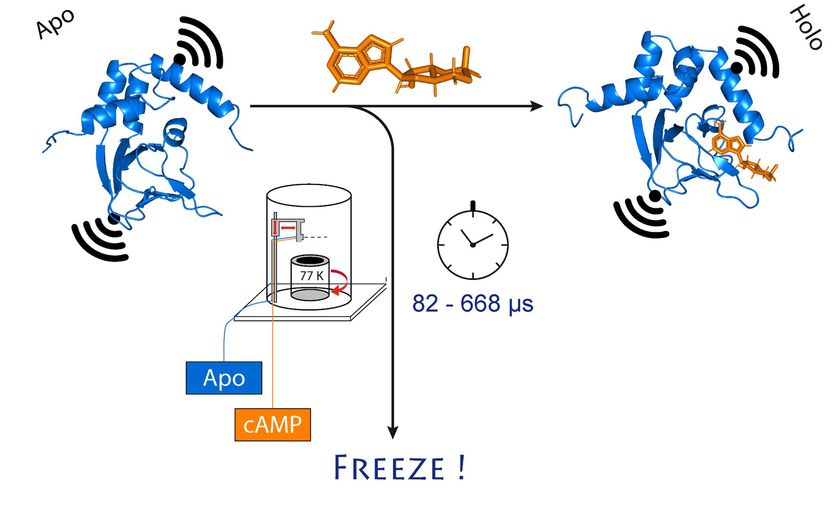
Una subunidad (azul) del canal de iones de potasio cambia su disposición cuando se une el AMPc (naranja). Esto también cambia la distancia entre los dos imanes moleculares unidos al canal (negro).
© Tobias Hett / University of Bonn and research center caesar
Si se quiere saber cómo es la estructura espacial de una biomolécula, se dispone de un formidable arsenal de herramientas. Las más populares son la microscopía electrónica y la difracción de rayos X, que pueden revelar hasta los detalles más pequeños de una proteína. Sin embargo, una limitación importante de estos métodos es que suelen ofrecer imágenes estáticas, que a menudo son insuficientes para comprender los procesos biomoleculares en términos mecanicistas precisos. Por ello, un objetivo a largo plazo de muchos grupos de investigación de todo el mundo ha sido seguir los movimientos dentro de una macromolécula como una proteína a lo largo del tiempo mientras realiza su trabajo, como en una película. Los grupos de investigación dirigidos por el Prof. Dr. Olav Schiemann, del Instituto de Química Física y Teórica de la Universidad de Bonn, y el Prof. Dr. Benjamin Kaupp, del centro de investigación Caesar de la Sociedad Max Planck, están ahora un paso más cerca de lograr este objetivo.
Han elegido un canal iónico para su investigación. Se trata de una proteína que forma minúsculos poros en la membrana celular que son permeables a las partículas cargadas llamadas iones. "Este canal está normalmente cerrado", explica Schiemann. "Sólo se abre cuando un mensajero celular, llamado AMPc, se une a él. Queríamos saber cómo funciona exactamente este proceso".
Mini imanes para medir distancias
Para ello, los investigadores mezclaron primero la proteína del canal y el AMPc y luego congelaron rápidamente la solución. En el estado congelado, la estructura de la proteína puede ahora ser analizada. Para que su método funcione, han colocado electroimanes moleculares en dos puntos del canal. La distancia entre estos imanes puede determinarse con una precisión de unos pocos Angstrom (diez mil millonésimas de milímetro) mediante un sofisticado método llamado PELDOR, que funciona como una regla molecular. En los últimos años, el grupo de Schiemann ha perfeccionado y mejorado considerablemente este método.
"Sin embargo, esto sólo nos da una imagen estática de la unión del AMPc al canal iónico", dice Schiemann. "Por lo tanto, repetimos el proceso de congelación en diferentes momentos después de mezclar las dos moléculas. Esto permitió reconstruir los movimientos de la proteína tras la unión del AMPc, como si se tratara de una película, que también se compone de una secuencia de imágenes".
En el centro de este procedimiento se encuentra un sofisticado método que permite mezclar y congelar las muestras muy rápidamente en un momento preciso. La técnica, denominada "hipercongelación de microsegundos" (abreviada MHQ), se desarrolló originalmente en la Universidad de Delft, pero luego cayó en desuso. El grupo de Kaupp la redescubrió y la perfeccionó de forma decisiva.
"En el dispositivo MHQ, la molécula de AMPc y el canal iónico se mezclan a velocidad ultrarrápida", explica Kaupp. "A continuación, la mezcla se dispara como un chorro muy fino sobre un cilindro metálico muy frío a -190 °C, que gira 7.000 veces por minuto. Resultó especialmente difícil transferir las muestras congeladas para la medición PELDOR desde la placa metálica a tubos de vidrio finos, y mantenerlas congeladas mientras tanto. Tuvimos que diseñar y construir herramientas especiales para ello".
Congelación en 82 millonésimas de segundo
Todo el proceso de mezcla y congelación dura sólo 82 microsegundos (un microsegundo equivale a una millonésima de segundo). "Esto nos permite visualizar cambios muy rápidos en la estructura espacial de las proteínas", explica Tobias Hett, uno de los dos estudiantes de doctorado que han contribuido significativamente al éxito. La ventaja del método es su combinación de alta resolución espacial y temporal. "Esto representa un gran paso adelante en el estudio de los procesos dinámicos en las biomoléculas", subraya Kaupp.
Los investigadores planean ahora utilizar su método para observar más de cerca otras biomoléculas. Esperan obtener nuevos conocimientos, por ejemplo, sobre el funcionamiento de las enzimas y los ácidos nucleicos. La importancia de estos conocimientos queda ilustrada por el reciente aumento de la investigación estructural sobre el coronavirus 2 del SARS en todo el mundo: la llamada proteína pico del virus también sufre un cambio estructural cuando se infectan las células humanas. El esclarecimiento de este mecanismo proporcionará una valiosa información sobre cómo atacar el mecanismo de infección con nuevos fármacos.
La preparación de las muestras, la ejecución experimental y el análisis de los datos son muy complejos. Por ello, los resultados del estudio reflejan también una fructífera cooperación científica con los investigadores dirigidos por el Prof. Dr. Helmut Grubmüller, del Instituto Max Planck de Química Biofísica de Gotinga, y el Prof. Dr. Heinz-Jürgen Steinhoff, de la Universidad de Osnabrück.
Nota: Este artículo ha sido traducido utilizando un sistema informático sin intervención humana. LUMITOS ofrece estas traducciones automáticas para presentar una gama más amplia de noticias de actualidad. Como este artículo ha sido traducido con traducción automática, es posible que contenga errores de vocabulario, sintaxis o gramática. El artículo original en Inglés se puede encontrar aquí.
Publicación original
Más noticias del departamento ciencias