El nuevo algoritmo agudiza el enfoque de los microscopios más poderosos del mundo
Los científicos desarrollan una técnica que mejora la resolución de la microscopía crioelectrónica
Todos hemos visto ese momento en un programa de TV de la policía donde un detective está revisando un granulado, material de seguridad de baja resolución, ve a una persona de interés en la cinta, y despreocupadamente pide a un técnico del CSI que "mejore eso". Unos pocos clics en el teclado después, y voila - tienen una perfecta y clara imagen de la cara del sospechoso. Esto, por supuesto, no funciona en el mundo real, como a muchos críticos de cine y gente en Internet les gusta señalar.
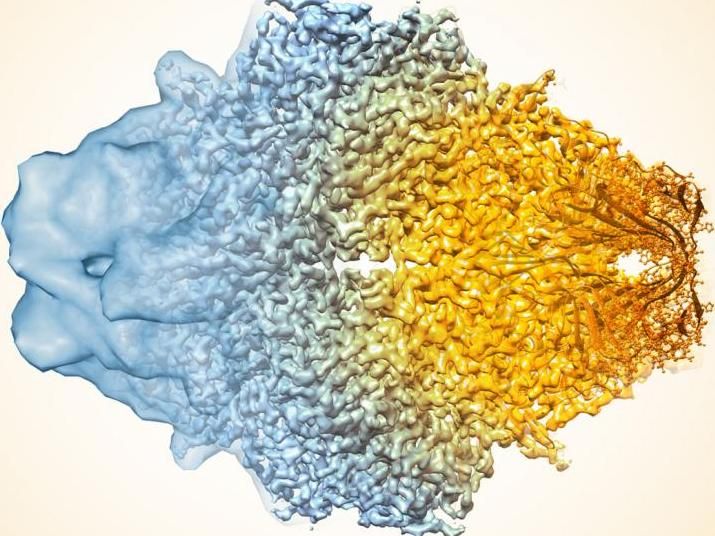
Una imagen compuesta de la enzima lactasa mostrando cómo la resolución de la crio-EM ha mejorado dramáticamente en los últimos años. Imágenes más antiguas a la izquierda, más recientes a la derecha.
Veronica Falconieri/National Cancer Institute
Sin embargo, los científicos de la vida real han desarrollado recientemente una verdadera herramienta de "mejora": una que mejora la resolución y la precisión de los poderosos microscopios que se utilizan para revelar conocimientos de biología y medicina.
En un estudio publicado en Nature Methods, un equipo multiinstitucional dirigido por Tom Terwilliger del Consorcio de Nuevo México e integrado por investigadores del Laboratorio Nacional Lawrence Berkeley (Berkeley Lab) demuestra cómo un nuevo algoritmo informático mejora la calidad de los mapas de estructura molecular tridimensionales generados con la microscopía crioelectrónica (crioEM).
Durante décadas, estos mapas de crio-mecánica, generados mediante la toma de muchas imágenes de microscopía y la aplicación de software de procesamiento de imágenes, han sido una herramienta crucial para los investigadores que buscan aprender cómo funcionan las moléculas dentro de los animales, las plantas, los microbios y los virus. Y en los últimos años, la tecnología crio-ME ha avanzado hasta el punto de que puede producir estructuras con una resolución a nivel atómico para muchos tipos de moléculas. Sin embargo, en algunas situaciones, incluso los más sofisticados métodos de crio-ME siguen generando mapas con menor resolución y mayor incertidumbre que la necesaria para conocer los detalles de las reacciones químicas complejas.
"En biología, ganamos mucho conociendo la estructura de una molécula", dijo el coautor del estudio Paul Adams, Director de la División de Biofísica Molecular y Bioimágenes Integradas del Laboratorio de Berkeley. "Las mejoras que vemos con este algoritmo facilitarán a los investigadores la determinación de modelos estructurales atomísticos a partir de datos de crio-microscopía electrónica. Esto es particularmente importante para modelar moléculas biológicas muy importantes, como las que participan en la transcripción y traducción del código genético, que a menudo sólo se ven en mapas de menor resolución debido a sus grandes y complejas estructuras de unidades múltiples".
El algoritmo agudiza los mapas moleculares filtrando los datos sobre la base de los conocimientos existentes sobre el aspecto de las moléculas y la mejor manera de estimar y eliminar el ruido (datos no deseados e irrelevantes) en los datos de microscopía. Anteriormente se había utilizado un enfoque con la misma base teórica para mejorar los mapas de estructuras generados a partir de la cristalografía de rayos X, y los científicos han propuesto su utilización en la crio-ME anteriormente. Pero, según Adams, nadie había sido capaz de mostrar pruebas definitivas de que funcionara para la crio-ME hasta ahora.
El equipo -compuesto por científicos del Consorcio de Nuevo México, el Laboratorio Nacional de Los Álamos, el Colegio de Medicina Baylor, la Universidad de Cambridge y el Laboratorio de Berkeley- aplicó por primera vez el algoritmo a un mapa disponible al público de la proteína humana apoferritina que se sabe que tiene una resolución de 3,1 angstroms (un angstromo es igual a una 10 mil millonésima parte de un metro; como referencia, se estima que el diámetro de un átomo de carbono es de 2 angstroms). Luego compararon su versión mejorada con otro mapa de referencia de apoferritina disponible públicamente con una resolución de 1,8 angstroms, y encontraron una mejor correlación entre ambos.
A continuación, el equipo utilizó su enfoque en 104 conjuntos de datos de mapas del Banco de Datos de Microscopía Electrónica. Para una gran proporción de estos conjuntos de mapas, el algoritmo mejoró la correlación entre el mapa experimental y la estructura atómica conocida, y aumentó la visibilidad de los detalles.
Los autores señalan que los claros beneficios del algoritmo en la revelación de detalles importantes en los datos, combinados con su facilidad de uso - es un análisis automatizado que puede realizarse en un procesador portátil - probablemente lo convierta en una parte estándar del flujo de trabajo de la crio-ME en marcha. De hecho, Adams ya ha añadido el código fuente del algoritmo a la suite de software Phenix, un paquete popular para la solución automatizada de estructura macromolecular para la que lidera el equipo de desarrollo.
Esta investigación fue parte de los continuos esfuerzos del Laboratorio de Berkeley para avanzar en las capacidades de la tecnología de crio-EM y para ser pioneros en su uso para los descubrimientos de la ciencia básica. Muchos de los inventos revolucionarios que permitieron el desarrollo de la crio-ME y que más tarde la llevaron a su excepcional resolución actual, han involucrado a los científicos del Laboratorio de Berkeley.
Nota: Este artículo ha sido traducido utilizando un sistema informático sin intervención humana. LUMITOS ofrece estas traducciones automáticas para presentar una gama más amplia de noticias de actualidad. Como este artículo ha sido traducido con traducción automática, es posible que contenga errores de vocabulario, sintaxis o gramática. El artículo original en Inglés se puede encontrar aquí.
Publicación original
Más noticias del departamento ciencias




















Estos productos pueden interesarle

FLUOVIEW FV4000 de EVIDENT
Imágenes revolucionarias con FLUOVIEW FV4000: escaneado láser confoca
Utilice el procesamiento de imágenes basado en IA y la innovadora tecnología de detectores

JEOL CRYO ARM de JEOL
Cryo-TEM: adquisición de datos rápida y estable para biomuestras
Mayor eficiencia en biología estructural con el sistema automatizado de carga de muestras

alpha300 R de WITec
Microscopio de imágenes Raman en 3D: Nano-analítica correlativa de alta resolución
Espectroscopia Raman de alta calidad