Bristol es pionera en el uso de la RV para el diseño de nuevos medicamentos
Anuncios



Los investigadores de la Universidad de Bristol son pioneros en el uso de la realidad virtual (RV) como herramienta para diseñar la próxima generación de tratamientos contra las drogas.

El usuario interactúa con una proteína en la RV.
University of Bristol
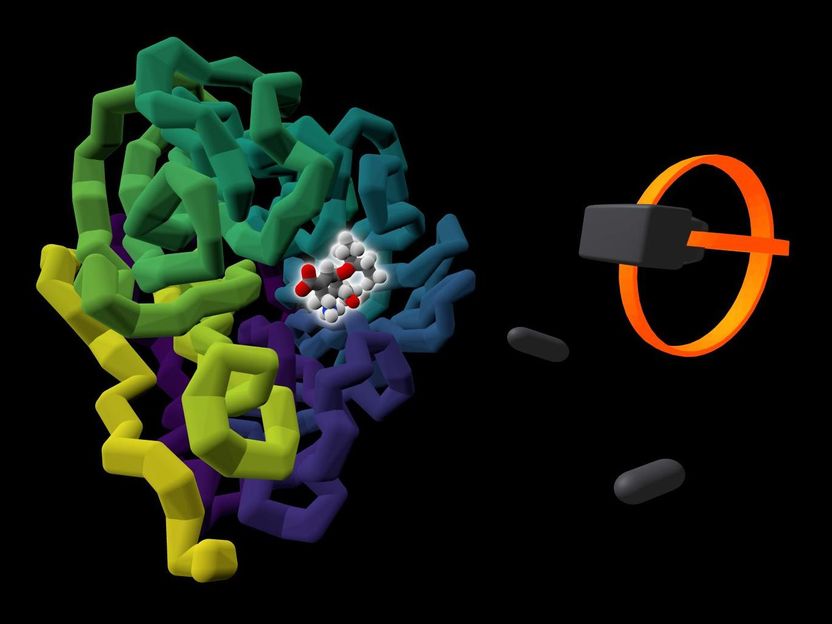
El medicamento contra la gripe Tamiflu, disponible en el mercado (resaltado por un brillo), se muestra unido a la proteína viral a la que se dirige, la neuraminidasa (de color entre púrpura y amarillo). Junto a la proteína se presenta una representación de los auriculares y controladores de RV del usuario.
University of Bristol

Los hallazgos, publicados en la revista PLOS ONE, describen cómo los investigadores utilizaron la RV para entender cómo funcionan los medicamentos comunes a nivel molecular.
Muchas drogas son pequeñas moléculas, y descubrir nuevas drogas implica encontrar moléculas que se unen a objetivos biológicos como las proteínas.
En el estudio, los usuarios fueron capaces de utilizar la RV para "meterse" en las proteínas y manipularlas, y las drogas que se unen a ellas, en detalle atómico, utilizando simulaciones interactivas de dinámica molecular en la RV (iMD-VR).
Usando este enfoque de iMD-VR, los investigadores 'acoplaron' las moléculas de la droga a las proteínas y fueron capaces de predecir con precisión cómo se unen las drogas. Entre los sistemas estudiados se encontraban los fármacos para la gripe y el VIH.
El profesor Adrian Mulholland, del Centro de Química Computacional de la Universidad de Bristol, y codirector del trabajo, dijo: "Muchos medicamentos funcionan uniéndose a las proteínas y deteniendo su funcionamiento. Por ejemplo, al unirse a una proteína de un virus determinado, una droga puede impedir que el virus se reproduzca.
"Para unirse bien, una droga de molécula pequeña necesita encajar bien en la proteína. Una parte importante del descubrimiento de drogas es encontrar pequeñas moléculas que se unen firmemente a proteínas específicas, y entender qué las hace unirse firmemente, lo que ayuda a diseñar mejores drogas.
"Para diseñar nuevas terapias, los investigadores necesitan entender cómo las moléculas de los fármacos encajan en sus objetivos biológicos. Para ello, utilizamos la RV para representarlas como objetos completamente tridimensionales. Los usuarios pueden entonces encajar una droga dentro del 'ojo de cerradura' de un sitio de unión de proteínas para descubrir cómo encajan."
En el estudio, los usuarios se fijaron la tarea de unir los medicamentos a objetivos de proteínas como la neuraminidasa de la gripe y la proteasa del VIH.
Las pruebas mostraron que los usuarios fueron capaces de predecir correctamente cómo las drogas se unen a sus objetivos de proteínas. Al introducir la droga en la proteína, pudieron construir estructuras muy similares a las estructuras de los complejos de drogas encontrados en los experimentos.
Incluso los no expertos fueron capaces de acoplar las drogas a las proteínas de manera efectiva. Esto demuestra que la RV interactiva puede ser utilizada para predecir con precisión cómo se unen los nuevos medicamentos potenciales a sus objetivos.
El estudio muestra cómo la RV puede utilizarse eficazmente en el diseño de drogas basadas en estructuras, incluso por personas no expertas. Utiliza equipos de RV fácilmente disponibles y un marco de software de código abierto, por lo que puede ser aplicado por cualquiera.
El profesor Mulholland añadió: "Un aspecto importante del trabajo es que las drogas, y sus blancos proteínicos, son totalmente flexibles: modelamos sus cambios estructurales y su dinámica, y los usuarios pueden manipularlos de forma interactiva para descubrir cómo interactúan las drogas con sus blancos biológicos. Esta es una forma realmente excitante y poderosa de modelar la unión de las drogas. Hemos demostrado en este trabajo que da resultados precisos. Estas herramientas serán útiles en el diseño y desarrollo de nuevas drogas".
El Dr. David Glowacki, Investigador Principal de la Sociedad Real en la Escuela de Química y el Departamento de Ciencias Informáticas de Bristol, dijo: "Nuestros resultados muestran que es posible desatar y volver a unir las drogas de los objetivos de las proteínas en una escala de tiempo de simulación significativamente más corta que la escala de tiempo de eventos similares observados usando motores de dinámica molecular no interactiva.
"También es importante señalar que los eventos de desacoplamiento y reacoplamiento completos generados usando iMD-VR fueron logrados por los usuarios en menos de cinco minutos de tiempo real.
"Cuando los usuarios no expertos tenían rastros de átomos que les mostraban la postura correcta, todos los participantes pudieron establecer una postura de acoplamiento lo suficientemente cercana a la estructura de partida como para ser considerada científicamente como reacoplada.
"Donde no había átomos de rastro, las posturas de unión tenían, comprensiblemente, más variación, pero los usuarios todavía podían estar dentro del mismo rango de la posición de unión aceptada para los tres sistemas. Estos resultados se lograron en una sola sesión de capacitación de una hora de duración con cada participante, lo que demuestra la utilidad de este marco de RV".
Nota: Este artículo ha sido traducido utilizando un sistema informático sin intervención humana. LUMITOS ofrece estas traducciones automáticas para presentar una gama más amplia de noticias de actualidad. Como este artículo ha sido traducido con traducción automática, es posible que contenga errores de vocabulario, sintaxis o gramática. El artículo original en Inglés se puede encontrar aquí.
Publicación original
H. Deeds, R. Walters, S. Hare, M. O’Connor, A. Mulholland and D. Glowacki; "Interactive molecular dynamics in virtual reality for accurate flexible protein-ligand docking"; PLOS One


Más noticias del departamento ciencias





















Noticias más leídas










Más noticias de nuestros otros portales



Contenido visto recientemente
Un enfoque innovador de la unión de células podría ayudar a entender las enfermedades

Hallado un posible marcador tumoral para el desarrollo del carcinoma hepatocelular - Las células cancerosas dejan una "huella" en las células asesinas naturales