Entender cómo afectan las mutaciones a las enfermedades
Nuevo marco para estudiar las enfermedades poligénicas
El código genético humano está totalmente cartografiado, lo que proporciona a los científicos un plano del ADN para identificar las regiones genómicas y sus variaciones responsables de enfermedades. Las herramientas estadísticas tradicionales localizan eficazmente estas "agujas en el pajar" genéticas, pero se enfrentan a dificultades para comprender cuántos genes contribuyen a las enfermedades, como ocurre en la diabetes o la esquizofrenia.
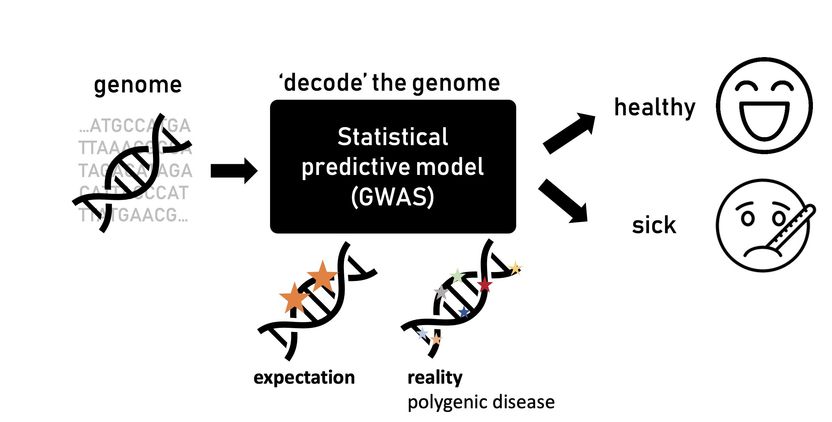
Los modelos tradicionales de GWAS, similares a una "caja negra", utilizan la estadística para predecir el riesgo de enfermedad a partir de los genomas. La mayoría de las enfermedades son poligénicas, es decir, muchos lugares del genoma contribuyen al riesgo de enfermedad, lo que dificulta la identificación de los mecanismos biológicos subyacentes.
Natália Ružičková / ISTA
Muchos modelos y algoritmos estadísticos utilizados por los científicos pueden imaginarse como una "caja negra". Estos modelos son herramientas poderosas que dan predicciones precisas, pero su funcionamiento interno no es fácilmente interpretable o comprensible. En una era dominada por el aprendizaje profundo, en la que se puede procesar una cantidad de datos cada vez mayor, Natália Ružičková, física y estudiante de doctorado en el Instituto de Ciencia y Tecnología de Austria (ISTA), optó por dar un paso atrás. Al menos en el contexto del análisis de datos genómicos.
Junto con Michal Hledík, recién graduado del ISTA, y el profesor Gašper Tkačik, Ružičková propuso ahora un modelo que podría ayudar a analizar las "enfermedades poligénicas", en las que muchas regiones del genoma contribuyen a un mal funcionamiento. Además, el modelo sirve para entender por qué las regiones genómicas identificadas contribuyen a estas enfermedades. Para ello combinan el análisis genómico más avanzado con conocimientos fundamentales de biología. Los resultados se publican en PNAS.
Descodificación del genoma humano
En 1990 se puso en marcha el Proyecto Genoma Humano para descifrar completamente el ADN humano, la huella genética que define a los seres humanos. En 2003, cuando se completó el proyecto, se allanó el camino para numerosos avances en ciencia, medicina y tecnología. Al descifrar el código genético humano, los científicos esperaban conocer mejor las enfermedades relacionadas con mutaciones y variaciones específicas de este guión genético. Dado que el genoma humano comprende aproximadamente 20.000 genes y aún más pares de bases -las letras del alfabeto azul-, se hizo esencial una gran potencia estadística. Esto condujo al desarrollo de los llamados "estudios de asociación de todo el genoma" (GWAS).
Los GWAS abordan la cuestión identificando variantes genéticas potencialmente vinculadas a rasgos del organismo como la estatura. Y lo que es más importante, también incluyen la propensión a diversas enfermedades. Para ello, el principio estadístico subyacente es bastante sencillo: los participantes se dividen en dos grupos: individuos sanos y enfermos. A continuación se analiza su ADN para detectar variaciones -cambios en su genoma- que son más prominentes en los afectados por la enfermedad.
Una interacción de genes
Cuando surgieron los estudios de asociación del genoma completo, los científicos esperaban encontrar sólo unas pocas mutaciones en genes conocidos relacionados con una enfermedad que explicaran la diferencia entre individuos sanos y enfermos. La verdad, sin embargo, es mucho más complicada. "A veces hay cientos o miles de mutaciones relacionadas con una enfermedad concreta", explica Ružičková. "Fue una revelación sorprendente y chocaba con la comprensión de la biología que teníamos".
Individualmente, cada mutación tiene un impacto o contribución mínimos al riesgo de desarrollar una enfermedad. Sin embargo, colectivamente, pueden explicar mejor, aunque no totalmente, por qué algunos individuos desarrollan la enfermedad. Tales enfermedades se denominan "poligénicas". Por ejemplo, la diabetes tipo 2 es poligénica, porque no puede atribuirse a un único gen, sino que implica cientos de mutaciones. Algunas de estas mutaciones afectan a la producción de insulina, a su acción o al metabolismo de la glucosa, mientras que la mayoría se localizan en regiones genómicas no relacionadas previamente con la diabetes o con funciones biológicas desconocidas.
El modelo omnigénico
En 2017, Evan A. Boyle y sus colegas de la Universidad de Stanford propusieron un nuevo marco conceptual llamado "modelo omnigénico". Propusieron una explicación de por qué tantos genes contribuyen a las enfermedades: las células poseen redes reguladoras que vinculan genes con diversas funciones.
"Como los genes están interconectados, una mutación en un gen puede afectar a otros, ya que el efecto mutacional se propaga por la red reguladora", explica Ružičková. Debido a estas redes, muchos genes del sistema regulador acaban contribuyendo a una enfermedad. Sin embargo, hasta ahora este modelo no se había formulado matemáticamente y seguía siendo una hipótesis conceptual difícil de probar. En su último trabajo, Ružičková y sus colegas introducen una nueva formalización matemática basada en el modelo omnigénico denominada "modelo omnigénico cuantitativo" (QOM).
Combinación de estadística y biología
Para demostrar el potencial del nuevo modelo, necesitaban aplicar el marco a un sistema biológico bien caracterizado. Eligieron el modelo habitual de levadura de laboratorio Saccharomyces cerevisiae, más conocida como levadura de cerveza o levadura de panadería. Se trata de un eucariota unicelular, lo que significa que su estructura celular es similar a la de organismos complejos como el ser humano. "En la levadura conocemos bastante bien cómo se estructuran las redes reguladoras que interconectan los genes", afirma Ružičková.
Utilizando su modelo, los científicos predijeron los niveles de expresión génica -la intensidad de la actividad génica, que indica cuánta información del ADN se utiliza activamente- y cómo se propagan las mutaciones a través de la red reguladora de la levadura. Las predicciones fueron muy eficaces: El modelo no sólo identificaba los genes relevantes, sino que también podía señalar claramente qué mutación contribuía con mayor probabilidad a un resultado específico.
Las piezas del rompecabezas de las enfermedades poligénicas
El objetivo de los científicos no era superar al GWAS estándar en rendimiento predictivo, sino ir en una dirección diferente haciendo que el modelo fuera interpretable. Mientras que un modelo GWAS estándar funciona como una "caja negra" que ofrece una relación estadística de la frecuencia con la que una mutación concreta se relaciona con una enfermedad, el nuevo modelo también proporciona un mecanismo causal de cadena de acontecimientos que explica cómo esa mutación puede provocar una enfermedad.
En medicina, comprender el contexto biológico y esas vías causales tiene enormes implicaciones para encontrar nuevas opciones terapéuticas. Aunque de momento el modelo está lejos de cualquier aplicación médica, muestra potencial, sobre todo para conocer mejor las enfermedades poligénicas. "Si se tienen suficientes conocimientos sobre las redes reguladoras, se podrían construir modelos similares también para otros organismos. Hemos estudiado la expresión génica en la levadura, lo que no es más que un primer paso y una prueba de principio. Ahora que entendemos lo que es posible, se puede empezar a pensar en aplicaciones a la genética humana", dice Ružičková.
Nota: Este artículo ha sido traducido utilizando un sistema informático sin intervención humana. LUMITOS ofrece estas traducciones automáticas para presentar una gama más amplia de noticias de actualidad. Como este artículo ha sido traducido con traducción automática, es posible que contenga errores de vocabulario, sintaxis o gramática. El artículo original en Inglés se puede encontrar aquí.
Publicación original
Más noticias del departamento ciencias








































