Mapas en 3D de la actividad de los genes
Anuncios



Un modelo computarizado tridimensional permite a los científicos determinar rápidamente qué genes están activos en qué células y su ubicación precisa dentro de un órgano. Un equipo dirigido por Nikolaus Rajewsky, de Berlín, y Nir Friedman, de Jerusalén, ha publicado el nuevo método y sus conocimientos adquiridos en Nature.

NovoSpaRc permite un rompecabezas tridimensional de expresión génica.
© Lior Friedman
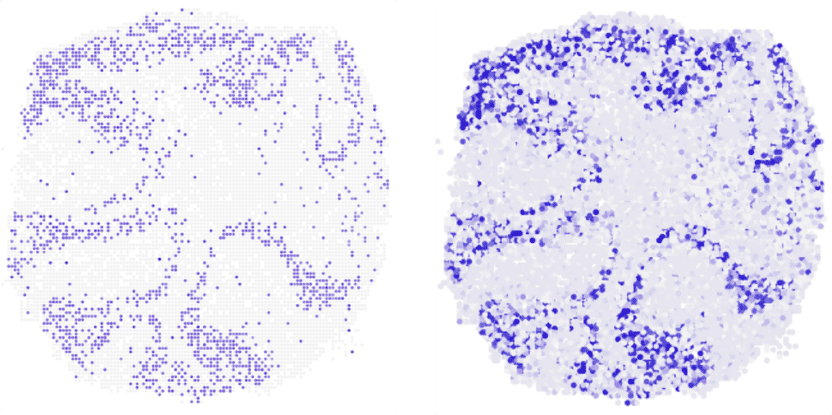
¿En qué parte del cerebelo del ratón se transcribe el gen Neurod1? La sección vertical muestra que novoSpaRc (derecha) es capaz de responder a esta pregunta de forma muy fiable sobre la base de los datos de secuenciación de una sola célula disponibles. A la izquierda para comparar el resultado de los experimentos.
© AG N. Rajewsky, MDC

El profesor Nikolaus Rajewsky es un visionario: quiere entender exactamente lo que sucede en las células humanas durante la progresión de la enfermedad, con el objetivo de poder reconocer y tratar los primeros cambios celulares. "Esto nos obliga no sólo a descifrar la actividad del genoma en células individuales, sino también a rastrearlo espacialmente dentro de un órgano", explica el director científico del Instituto de Biología de Sistemas Médicos de Berlín (BIMSB) en el Centro Max Delbrück de Medicina Molecular (MDC) de Berlín. Por ejemplo, la disposición espacial de las células inmunitarias en el cáncer ("microambiente") es extremadamente importante para diagnosticar la enfermedad con precisión y seleccionar la terapia óptima. "En general, carecemos de un enfoque sistemático para capturar molecularmente y entender la (pato-)fisiología de un tejido."
Mapas para tipos de tejido muy diferentes
Rajewsky ha dado un gran paso hacia su objetivo con un nuevo e importante estudio que ha sido publicado en la revista científica Nature. Junto con el profesor Nir Friedman de la Universidad Hebrea de Jerusalén, el Dr. Mor Nitzan de la Universidad de Harvard en Cambridge, EE.UU., y el Dr. Nikos Karaiskos, jefe de proyecto de su propio grupo de investigación sobre "Biología de Sistemas de Elementos Reguladores de Genes", los científicos han logrado utilizar un algoritmo especial para crear un mapa espacial de expresión génica para células individuales en tipos de tejido muy diferentes: en el hígado y en el epitelio intestinal de mamíferos, así como en embriones de moscas de la fruta y peces cebra, en partes del cerebelo, y en el riñón. "A veces, la ciencia puramente teórica es suficiente para publicar en una revista científica de alto nivel; creo que esto ocurrirá con mayor frecuencia en el futuro. Tenemos que invertir mucho más en el aprendizaje automático y la inteligencia artificial", dice Nikolaus Rajewsky.
"Utilizando estos mapas generados por ordenador, ahora podemos rastrear con precisión si un gen específico está activo o no en las células de una parte del tejido", explica Karaiskos, un físico teórico y bioinformático que desarrolló el algoritmo junto con Mor Nitzan. "Esto no hubiera sido posible en esta forma sin nuestro modelo, que hemos llamado'novoSpaRc.'"
La información espacial se perdió anteriormente
Sólo en los últimos años los investigadores han podido determinar, a gran escala y con gran precisión, la información que las células individuales de un órgano o tejido están obteniendo del genoma en un momento dado. Esto se debe a los nuevos métodos de secuenciación, por ejemplo, la secuenciación de ARN múltiplex, que permite analizar simultáneamente un gran número de moléculas de ARN. El ARN se produce en la célula cuando los genes se activan y las proteínas se forman a partir de sus planos. Rajewsky reconoció el potencial de la secuenciación unicelular desde el principio y la estableció en su laboratorio.
"Pero para que esta tecnología funcione, el tejido bajo investigación debe primero descomponerse en células individuales", explica Rajewsky. Este proceso hace que se pierda información valiosa: por ejemplo, la ubicación original en el tejido de la célula en particular cuya actividad genética ha sido decodificada genéticamente. Por lo tanto, Rajewsky y Friedmann buscaban una forma de utilizar los datos de la secuenciación unicelular para desarrollar un modelo matemático que pudiera calcular el patrón espacial de la expresión génica para todo el genoma, incluso en tejidos complejos.
Los equipos dirigidos por Rajewsky y el Dr. Robert Zinzen, que también trabaja en el BIMSB, ya lograron un primer avance hace dos años. En la revista científica Science, presentaron un modelo virtual de un embrión de mosca de la fruta. Mostraba qué genes estaban activos en qué células en una resolución espacial que nunca antes se había logrado. Este mapeo genético fue posible con la ayuda de 84 genes marcadores: los experimentos in situ habían determinado en qué parte del embrión en forma de huevo estaban activos estos genes en un momento determinado. Los investigadores confirmaron que su modelo trabajaba con otros complejos en experimentos sobre embriones vivos de mosca de la fruta.
Un rompecabezas con decenas de miles de piezas y colores
"En este modelo, sin embargo, reconstruimos la ubicación de cada célula individualmente", dijo Karaiskos. Fue uno de los primeros autores tanto del estudio "Science" como del actual estudio "Nature". "Esto fue posible porque tuvimos que tratar con un número considerablemente menor de células y genes. Esta vez, queríamos saber si podemos reconstruir tejidos complejos cuando apenas tenemos información previa. ¿Podemos aprender un principio sobre cómo se organiza y regula la expresión génica en tejidos complejos?" La suposición básica para el algoritmo era que cuando las células son vecinas, su actividad genética es más o menos parecida. Ellos recuperan más información similar de su genoma que las células que están más alejadas.
Para probar esta hipótesis, los investigadores utilizaron los datos existentes. Para el hígado, los riñones y el epitelio intestinal no hubo información adicional. El grupo había podido recolectar sólo unos pocos genes marcadores utilizando muestras de tejido reconstruido. En un caso, sólo había dos genes marcadores disponibles.
"Fue como armar un rompecabezas masivo con un gran número de colores diferentes, quizás 10.000 o más", explica Karaiskos, tratando de describir la difícil tarea a la que se enfrentó al calcular el modelo. "Si el rompecabezas se resuelve correctamente, todos estos colores dan como resultado una forma o patrón específico." Cada pieza del rompecabezas representa una sola célula del tejido bajo investigación, y cada color un gen activo que fue leído por una molécula de ARN.
El método funciona independientemente de la técnica de secuenciación
"Ahora tenemos un método que nos permite crear un modelo virtual del tejido bajo investigación sobre la base de los datos obtenidos de la secuenciación unicelular en el ordenador, independientemente del método de secuenciación que se haya utilizado", dice Karaiskos. "La información existente sobre la ubicación espacial de las células individuales puede ser introducida en el modelo, perfeccionándolo aún más." Con la ayuda de novoSpaRc, es posible determinar para cada gen conocido en qué parte del tejido el material genético está activo y se está traduciendo en una proteína.
Ahora, Karaiskos y sus colegas de BIMSB también se están enfocando en usar el modelo para rastrear e incluso predecir ciertos procesos de desarrollo en tejidos u organismos enteros. Sin embargo, el científico admite que puede haber algunos tejidos específicos que son incompatibles con el algoritmo novoSpaRc. Pero esto podría ser un desafío bienvenido, dice: Una oportunidad para probar su mano en un nuevo rompecabezas!
Nota: Este artículo ha sido traducido utilizando un sistema informático sin intervención humana. LUMITOS ofrece estas traducciones automáticas para presentar una gama más amplia de noticias de actualidad. Como este artículo ha sido traducido con traducción automática, es posible que contenga errores de vocabulario, sintaxis o gramática. El artículo original en Inglés se puede encontrar aquí.
Publicación original

Más noticias del departamento ciencias




































