Evolution bei der Wirkstoffsuche
Forscher des Leibniz-Instituts für Molekulare Pharmakologie (FMP) haben ein Verfahren entwickelt, mit dem sie Wirkstoffe direkt auf der Oberfläche von Proteinen herstellen und gleichzeitig ihre Wirksamkeit testen. Das könnte das Screening von Substanzen erheblich beschleunigen.
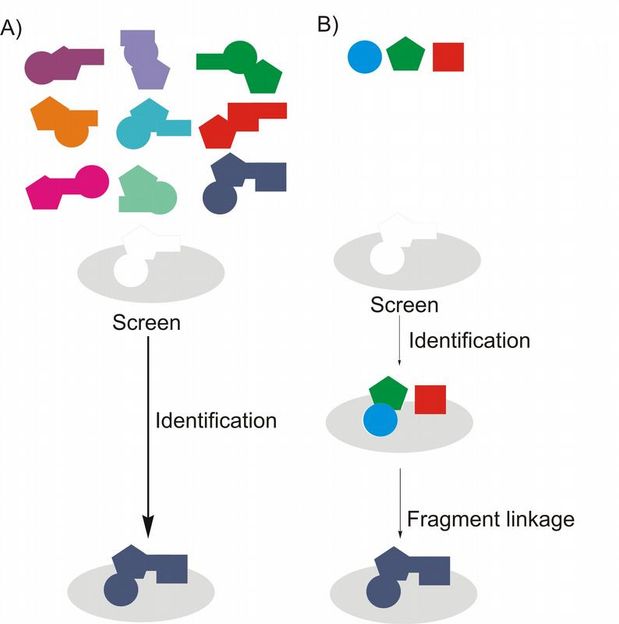
A) Herkömmliches Screeningverfahren mittels kombinatorischer Chemie und Hochdurchsatz-Screening: Eine Substanzbibliothek von Molekülen mit (nahezu) jeder möglichen Kombination wird synthetisiert und am Zielprotein getestet. B) Konzept der fragmentbasierten Leitstruktur: Die drei geometrischen Formen stehen für Fragmente, die an das Zielprotein binden. Diese können chemisch verbunden werden. Trotz kleinerer Substanzbibliotheken, kann so schneller die passende Leitstruktur gefunden werden.
FMP
Im Körper gibt es unzählige Proteine, die Ziele für Wirkstoffe sein können. Viele Proteine sind Enzyme, das heißt sie unterstützen chemische Reaktionen im Organismus. "Wenn wir solche Proteine an- und ausschalten können, ist dies der Schlüssel zur Behandlung und Diagnose von Krankheiten", sagt Jörg Rademann, Leiter der Abteilung Medizinische Chemie am FMP. Die Suche nach Substanzen, die wie ein Schlüssel ins Schloss der Enzyme passen, gleicht dabei aber der Suche nach der Stecknadel im Heuhaufen. Forscher wenden deshalb automatisierte Prozesse an, mit denen sie viele Millionen Substanzen durch biochemische Tests schleusen - sogenannte Hochdurchsatz-Screenings - immer in der Hoffnung auf den entscheidenden Treffer.
In solche Screenings steckt die Pharmaindustrie immer mehr Geld, gleichzeitig werden jedoch immer weniger neue Medikamente zugelassen. Woran liegt das? Laut einer Studie gibt es etwa zehn hoch acht synthetisierte Verbindungen, die überall auf der Welt durchgescreent werden. Wollte man zehn hoch dreißig Verbindungen herstellen, würde die Materie im ganzen Universum nicht ausreichen. Geht man von Berechnungen aus, wonach es zehn hoch dreiundsechzig Möglichkeiten gibt, wie ein Wirkstoff aufgebaut sein kann, wird klar, dass solche Screenings nicht zum Erfolg führen können.
Die Forscher des FMP setzen deshalb auf eine quasi evolutionäre Methode. Sie screenen zunächst Fragmente eines potenziellen Wirkstoffes, die in nur eine Bindungsstelle des Zielenzyms passen. Haben sie hier einen geeigneten Kandidaten gefunden, verwenden sie diesen als feste Größe und geben nun Fragmente hinzu, die in die zweite Bindungsstelle passen. Die beiden Fragmente sind jeweils mit zwei unterschiedlichen reaktiven Gruppen versehen, die auf der Oberfläche des Enzyms eine chemische Reaktion miteinander eingehen. Haben die Forscher für Bindungsstelle 2 ebenfalls einen optimalen Kandidaten gefunden, nehmen sie diesen als feste Größe und variieren nochmals die Fragmente für Bindungsstelle 1.
"Wenn wir auf diese Weise je einhundert Substanzen screenen, haben wir am Ende nicht zweihundert Möglichkeiten getestet, sondern das Produkt davon, also 10.000", sagt Marco Schmidt, der im Rahmen seiner Doktorarbeit mit an der Methode gearbeitet hat. Am Ende dieses Verfahrens, das die Forscher dynamisches Ligationsscreening nennen, steht dann eine Substanz, die genau in die beiden Bindungsstellen des Enzyms passt.
Auf diese Weise konnten die FMP-Forscher für die SARS-Coronavirus-Hauptprotease eine Substanz finden, die das Enzym im mikromolaren Bereich hemmt. "Unsere Methode erlaubt es, relativ rasch und mit geringem Aufwand niedermolekulare Moleküle zu identifizieren, die Leitstrukturen für neue Wirkstoffe sein könnten", sagt Rademann. Kleine Moleküle sind im Gegensatz zu langen Peptidstrukturen in der Lage, die Zellmembran zu durchdringen, außerdem sind sie im Körper länger stabil und können oral verabreicht werden. Sie sind deshalb die bevorzugten Strukturen bei der Wirkstoffentwicklung. Die Methode des FMP könnte vor allem für die Pharmaindustrie interessant sein, die Forscher haben sie deshalb auch als Patent angemeldet.
Die Forscher versuchen derzeit für weitere krankheitsrelevante Proteine Hemmstoffe zu finden, so etwa für die Caspase 3, die am Zelltod von Gehirnzellen nach einem Schlaganfall beteiligt ist und für Phosphatasen, die bei der Krebsentstehung eine Rolle spielen. Rademann betont, dass das FMP das Verfahren auch anderen Wissenschaftlern im Rahmen des Netzwerkes ChemBioNet zur Nutzung zur Verfügung stellt. "Gerade im Bereich der Wirkstoffsuche müssen Biologen und Chemiker besonders eng zusammenarbeiten, damit die Funktion der entwickelten Moleküle im biologischen Kontext untersucht und verstanden werden kann."
Originalveröffentlichung: Marco Florian Schmidt et al.; "Sensibilisierte Detektion inhibitorischer Fragmente und iterative Entwicklung nicht-peptidischer Proteaseinhibitoren durch dynamisches Ligationsscreening"; Angew. Chem. 2008, 120, 3319 -3323
Weitere News aus dem Ressort Wissenschaft


















Diese Produkte könnten Sie interessieren

Antibody Stabilizer von CANDOR Bioscience
Protein- und Antikörperstabilisierung leicht gemacht
Langzeitlagerung ohne Einfrieren – Einfache Anwendung, zuverlässiger Schutz

DynaPro NanoStar II von Wyatt Technology
NanoStar II: DLS und SLS mit Touch-Bedienung
Größe, Partikelkonzentration und mehr für Proteine, Viren und andere Biomoleküle


































