Neues Werkzeug zur Entschlüsselung der Evolutionsbiologie
Eine Forschungsgruppe der Max F. Perutz Laboratories, einem Joint Venture der Universität Wien und der Medizinischen Universität Wien, hat gemeinsam mit anderen Wissenschaftern aus Australien und Kanada ein neues bioinformatisches Werkzeug zum Vergleich von Genomdaten entwickelt. Das Programm „ModelFinder“ verwendet einen schnellen Algorithmus, der bisher nicht verfügbare Einsichten in die Evolution gibt.
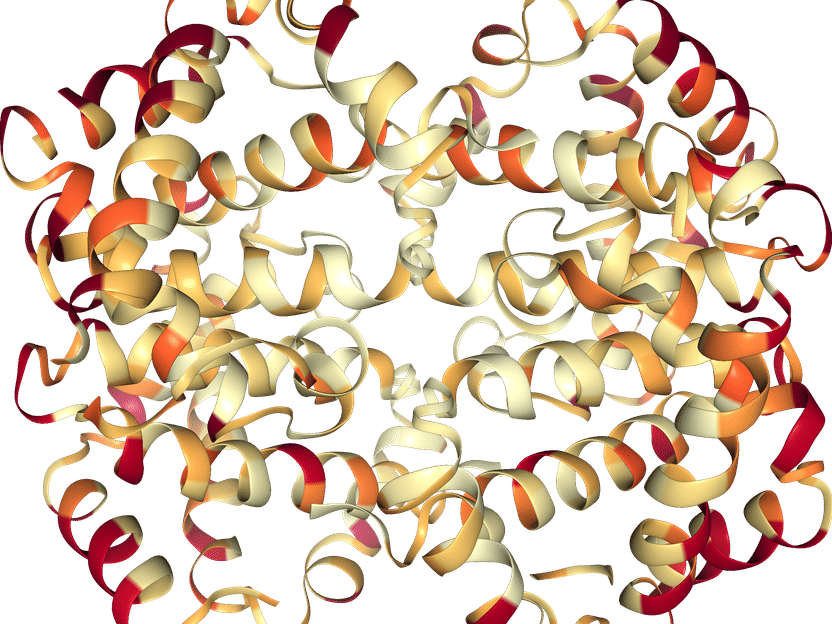
3D-Struktur des menschlichen Hämoglobins. Teile mit hoher (rot) oder niedriger (gelb) Evolutionsrate wurden durch ModelFinder ermittelt.
Copyright: Minh Quang Bui/Universität Wien
Die Evolution zu verstehen, ist ein Grundpfeiler der Biologie – tatsächlich ist Evolution die einzige Erklärung für die Vielfalt der Lebensformen auf unserem Planeten.
Anhand der Evolution von Proteinen kann erklärt werden, wie neue Spezies und Funktionen durch genetische Veränderungen entstehen konnten, wie Enzyme mit neuen Funktionen hergestellt werden können, oder auch wie ähnlich menschliche Genome zu jenen ihrer engsten Verwandten wie Gorillas oder Bonobos sind.

Sehr beliebte Tools sind bioinformatische Werkzeuge (computergestützte Methoden), mit denen sich Genomdaten vergleichen lassen. So können Wissenschafter spezifische Proteine vergleichen, die durch Kombinationen von zwanzig universellen Bausteinen – den Aminosäuren – gebildet werden.
Bis jetzt haben bioinformatische Werkzeuge für die Erforschung der Evolution von einzelnen Proteinen angenommen, dass die Geschwindigkeit, mit denen unterschiedliche Regionen von Proteinen evolvieren, durch eine statistische Verteilung, deren Form durch eine einzige Variable bestimmt wird, modelliert werden kann.
„Diese Annahme reflektiert allerdings nicht die Realität. Sie könnte zu einer großen Zahl von verzerrten phylogenetischen Resultaten geführt haben, die über die letzten beiden Jahrzehnte publiziert wurden“, fasst Minh Quang Bui, Co-Autor der Studie vom Center for Integrative Bioinformatics Vienna (CIBIV), die Grenzen der modernen Methoden zusammen.
Neuer Algorithmus gibt Einsichten in die Proteinevolution
Arndt von Haeseler, Gruppenleiter an den Max F. Perutz Laboratories (MFPL), und Lars Jermiin von der Australian National University konnten nun einen neuen Weg finden, um diese unterschiedlichen Evolutionsraten in bioinformatische Modelle miteinzubeziehen.
In der Fachwelt war es wohlbekannt, dass die gängige Methode die Komplexität der Proteinevolution möglicherweise nicht fassen könnte. Der Rechenaufwand von realistischeren Modellen war allerdings inakzeptabel hoch. „Wir haben nun einen schnellen Algorithmus entwickelt, der uns bis dato nicht verfügbare Einsichten in die Proteinevolution gibt – das neue Tool wird wahrscheinlich einen sehr großen Einfluss auf viele andere Forschungsgebiete haben, wie zum Beispiel auf die Evolution von Pathogenen und die Verbreitung von Agrarschädlingen“, sagt Lars Jermiin.
Das neue Programm „ModelFinder“ wird deutlich exaktere Schätzungen von evolutionären Prozessen ermöglichen. Das verbesserte Verständnis der Evolution hilft, jene Mechanismen, die für die große Diversität auf unserem Planeten verantwortlich sind, weiter zu enträtseln.
Originalveröffentlichung

Weitere News aus dem Ressort Wissenschaft


















Diese Produkte könnten Sie interessieren

Antibody Stabilizer von CANDOR Bioscience
Protein- und Antikörperstabilisierung leicht gemacht
Langzeitlagerung ohne Einfrieren – Einfache Anwendung, zuverlässiger Schutz

DynaPro NanoStar II von Wyatt Technology
NanoStar II: DLS und SLS mit Touch-Bedienung
Größe, Partikelkonzentration und mehr für Proteine, Viren und andere Biomoleküle

Meistgelesene News











Weitere News von unseren anderen Portalen






















Zuletzt betrachtete Inhalte
Vielfältige Nervenschäden durch Alkohol in der Schwangerschaft
LINNEMANN GmbH - Tübingen, Deutschland
