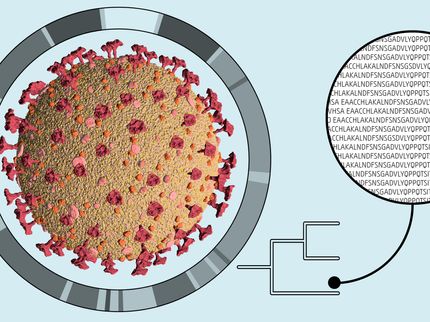
Neuartige Datenbank für Erbinformationen soll Forschung revolutionieren
Im Jahr 2000 verkündeten Wissenschaftler der Welt, das Erbgut eines Menschen komplett analysiert zu haben. Mit der daraus entstandenen Gen-Karte, Referenzgenom genannt, erforschen sie seitdem, wie Gene biologische Prozesse steuern. Inzwischen benötigt die Sequenzierung eines Genoms jedoch nur noch Stunden anstatt Jahre. Die Auswertung dieser Gesamtmenge an Gen-Daten ist der nächste Schritt, um Krankheiten wie Krebs besser behandeln zu können. Eine internationale Forschergemeinde arbeitet daher an einer neuartigen Datenbank, um diese umfangreichen Gen-Daten besser aufzubereiten. Mit dabei ist Tobias Marschall, Juniorprofessor am Zentrum für Bioinformatik der Saar-Uni.
„Stellen Sie sich ein riesiges Puzzle vor. Um es schnell zu vollenden, orientieren Sie sich an der Abbildung des fertigen Puzzles auf dem Karton“, erklärt Tobias Marschall die Funktion des Referenzgenoms. Am Zentrum für Bioinformatik entwickelt er neue Rechenverfahren, die das Gen-Puzzle nicht nur effizient lösen, sondern den Wissenschaftlern weitere Erkenntnisse liefern. Diese Automatisierung der Analyse ist notwendig, da die Menge an Gen-Daten sprunghaft angestiegen ist. Inzwischen nimmt die Sequenzierung eines Genoms, der Gesamtheit aller Erbinformationen eines Lebewesens oder Virus, nicht mehr Jahre in Anspruch, sondern dauert nur noch Stunden.
„Mit den Daten, die wir heute haben, können wir viel mehr leisten“, fasst Marschall die aktuelle Lage zusammen. Beispielsweise könne man die Unterschiede zwischen den Genomen von gesunden und erkrankten Menschen ermitteln und dann prüfen, ob diese statistisch signifikant seien. „Sie können die entscheidenden Ansatzpunkte bieten, um sicherzustellen, dass jeder Patient eine maßgeschneiderte Therapie bekommt“, erklärt Marschall. Jedoch gerade diese Art von Forschung ist mit dem aktuellen Referenzgenom schwierig. „Das Abbild eines fertigen Puzzles hilft beim Puzzeln nur, wenn die Ähnlichkeiten hoch sind“, so Marschall.
Zusammen mit einer Gruppe von hochkarätigen Forscherkollegen wie Professor Knut Reinert von der Freien Universität Berlin, der bereits bei der ersten Sequenzierung des Humangenoms mitgearbeitet hatte, plant er daher den Schritt weg von einem Referenzgenom hin zu einer Art intelligentem Gen-Netzwerk. Im Gegensatz zum bisherigen Referenzgenom soll es nicht nur die Informationen zu einem sequenzierten Genom bereitstellen, sondern die aller sequenzierten Genome. Mit Hilfe mathematischer Verfahren könnte dann nicht nur jede Kombination eines menschlichen Genoms berechnet werden, sondern ebenso die Erbinformation einer gesamten Bevölkerung, ein funktionales Genom, bei dem hinderliche Mutationen ausgeklammert werden, und sogar das maximale Genom, das alle bisher detektierten Sequenzen umfasst.
Die Wissenschaftler bezeichnen diese neuartige Referenzdarstellung als Pangenom und das dadurch definierte Forschungsgebiet als „computational pan-genomics“, was sich als „rechnergestützte, allumfassende Genomik” übersetzen lässt. Die dafür notwendigen Methoden und Algorithmen lassen sich nicht nur auf das menschliche Genom anwenden. Zusammen mit rund 60 Wissenschaftlern hat Marschall gerade den wissenschaftlichen Aufsatz „Computational pan-genomics: status, promises and challenges“ veröffentlicht. Darin definieren die Wissenschaftler sieben Forschungsgebiete. Neben Erbkrankheiten sind dies unter anderem Mikroben, Viren, Metagenomik, Pflanzen und Krebs.
Als Datenstruktur verwenden die Forscher Graphen und bedienen sich der dahinterstehenden, mathematischen Theorie. Als Graph bezeichnen Informatiker ein Modell, mit dessen Hilfe sich Objekte beschreiben lassen, die in Beziehung stehen. Anschauliche Beispiele sind U-Bahn-Pläne oder Familienstammbäume. „Das wird insbesondere uns Bioinformatiker fordern“, erklärt Marschall. Die effiziente Suche in Graphen sei viel aufwendiger als das bisherige Durchstöbern von Zeichenketten. Doch die Anstrengungen der Informatik alleine werden nicht ausreichen, glaubt Marschall. „Auch die Politik und die Gesellschaft ist gefordert. Nur so können wir die Erbinformationen aus allen Ländern der Welt für die Wissenschaft zugänglich machen. Die Aussicht, dadurch die Gesundheitsversorgung von Millionen von Menschen zu verbessern, sollte Motivation genug sein“, so Marschall.
Originalveröffentlichung
Weitere News aus dem Ressort Wissenschaft




















Meistgelesene News










Weitere News von unseren anderen Portalen






















Zuletzt betrachtete Inhalte

Was tun gegen einen Biss der Schwarzen Witwe? - Neue Antikörper entwickelt – mit weniger Nebenwirkungen und ohne Tierversuche
Wuschel
Kein Stau am Golgi-Apparat - Biochemiker zeichnen erstmals die Wanderwege von Ras-Proteinen in lebenden Zellen nach
Nocardiaceae
Bernhard_Grzimek
Selektion beeinflusst viele Gene für Übergewicht parallel - Max-Planck-Wissenschaftler entschlüsseln Gene für eine komplexe Eigenschaft