Algorithmen eröffnen Einblicke in die Zellentwicklung
Durch RNA-Sequenzierung können Forscher messen, welche Gene in jeder einzelnen Zelle einer Probe abgelesen werden. Eine neue statistische Methode erlaubt es, so aus einem Zellgemisch unterschiedliche Entwicklungsprozesse herauszulesen, die nebeneinander ablaufen. Das berichten Forscherinnen und Forscher des Helmholtz Zentrums München in Zusammenarbeit mit Kollegen der Technischen Universität München.
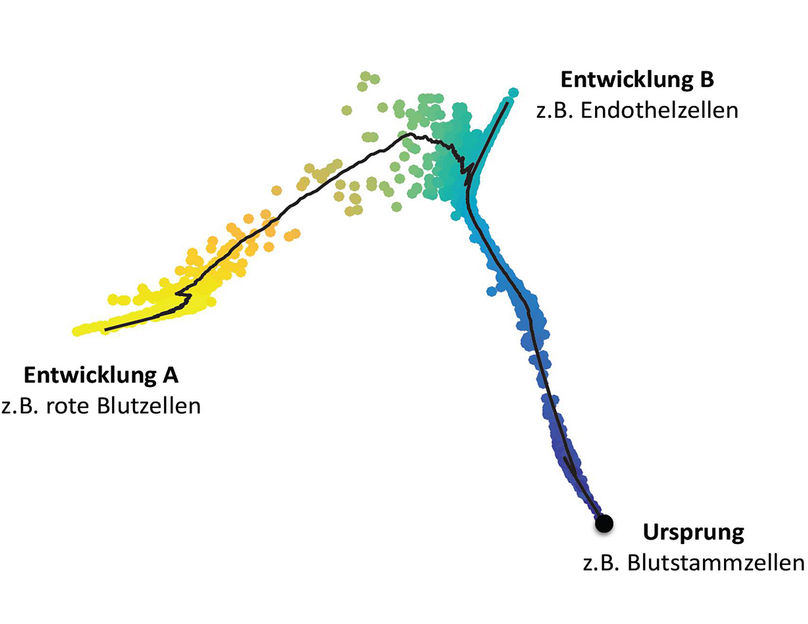
Entwicklung eines Verband von Blutstammzellen zu unterschiedlichen Zelltypen
HMGU
Die Zellbiologie befasst sich heutzutage oft nicht mehr nur mit statischen Zuständen, sondern möchte vielmehr die dynamische Entwicklung von Zellen verstehen. Ein Beispiel dafür ist die Bildung verschiedener Typen von Blutzellen aus ihren Vorläufern, den Blutstammzellen. Um zu verstehen, wie dieser Prozess genetisch gesteuert wird, analysieren Wissenschaftler, welche Gene abgelesen werden - das sogenannte Transkriptom.
„Dass wir heutzutage sogar das Transkriptom von Einzelzellen bestimmen können, ist für mich immer noch sehr erstaunlich“, sagt Erstautorin Laleh Haghverdi. „Vor allem, wenn man sich klar macht, dass eine typische Zelle nur wenige Billionstel Gramm RNA in sich trägt.“ Die Verfügbarkeit dieser Daten beginnt nun, viele Forschungsfelder zu revolutionieren, verlangt aber auch neue statistische Methoden, um sie richtig zu interpretieren. „So starten beispielsweise nie alle Zellen einer Probe gleichzeitig ihre Entwicklung und brauchen auch unterschiedlich lange. Daher haben wir es immer mit einem dynamischen Gemisch zu tun“, so die Doktorandin vom Institute of Computational Biology (ICB) am Helmholtz Zentrum München weiter. „Daraus die Abfolge von mehreren Schritten eines Prozesses zu konstruieren, ist enorm schwer, zumal die Zellen nur für eine Messung zur Verfügung stehen.“
Willkommen im Pseudozeitalter
Um also Entwicklungsprozesse aus der Messung eines einzigen Zeitpunkts, quasi einer Schnappschussmessung, zu entschlüsseln, entwickelten die Forscherinnen und Forscher um ICB-Direktor Prof. Dr. Dr. Fabian Theis einen Algorithmus namens Diffusion Pseudotime zur Interpretation von Einzelzell-Sequenzierungsdaten. Dieser ordnet Zellen auf einer virtuellen Zeitachse – der Pseudozeit – entlang derer sie kontinuierliche Veränderungen im Transkriptom aufweisen. Dadurch lässt sich rekonstruieren, welche Gene nacheinander abgelesen werden. So können die Forscher grafisch darstellen, wie sich die Entwicklungspfade unterschiedlicher Zelltypen verzweigen.
„Wir können beispielsweise zeigen, wie sich ein relativ einheitlicher Verband von Blutstammzellen zu unterschiedlichen Zelltypen entwickelt“, erklärt Studienleiter Theis. „Während die einen zu roten Blutkörperchen werden, differenzieren andere zu endothelartigen Zellen. Diese Schicksale können wir anhand der Transkriptom-Daten der Einzelzellen nachzeichnen.“ Zudem erhalten die Wissenschaftler Information darüber, welche Genschalter hinter den Entwicklungen stecken. Der relativ diffuse Mix von Zellen, die sich auf verschiedenen Etappen ihrer Entwicklung befanden, kann so also am Rechner entwirrt werden und erlaubt nach der Analyse ein klares Bild auf die ablaufenden Einzelschritte.
Doch wenn es nach den Forschern geht, soll das nur der Anfang gewesen sein, denn die Prozesse der Blutbildung sind schon relativ gut verstanden. Sie dienten nur als Testobjekt für die Leistungsfähigkeit der Methode. „Künftig wollen wir uns vor allem Prozesse anschauen, die bisher unverstanden oder möglicherweise noch gar nicht entdeckt sind“, so Theis.
Originalveröffentlichung
Weitere News aus dem Ressort Wissenschaft






















































