Aggressiver Bauchspeicheldrüsenkrebs: Welche Rolle spielen Entzündungssignale?
Bauchspeicheldrüsenkrebs ist eine der gefährlichsten Krebsarten überhaupt. Gegen Strahlen- und Chemotherapie ist er praktisch resistent und daher kaum zu behandeln. Darüber hinaus streut er früh in die Umgebung und wird oft spät entdeckt. Viele Patienten sterben wenige Monate nach der Diagnose. Warum Pankreaskrebs aber so aggressiv ist, lag lange im Dunkeln. Ein Team um Jens Siveke am Klinikum rechts der Isar der TU München hat sich auf die Spur der molekularen Veränderungen in den Krebszellen gemacht. Dabei gab es eine Überraschung: ein kleines Molekül, das Entzündungssignale in der Bauchspeicheldrüse reguliert, hemmt entscheidend die Tumorentstehung. Seine Rolle war bisher nicht bekannt.
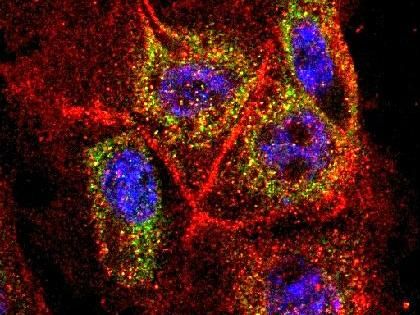
Krebszellen aus der Bauchspeicheldrüse unter dem Fluoreszenzmikroskop.
Jens Siveke, Klinikum rechts der Isar, München
In Deutschland erkranken jedes Jahr rund 15.000 Menschen an Bauchspeicheldrüsenkrebs – und fast alle sterben innerhalb eines Jahres daran. Zu wenig ist bisher darüber bekannt, was genau die Zellen der Bauchspeicheldrüse zu ihrem unkontrollierten Wachstum anregt. Allerdings zeigte sich bei Untersuchungen von Krebszellen aus der Bauchspeicheldrüse in fast allen Fällen eine Übereinstimmung: Ein bestimmtes Gen war mutiert, das KRAS-Onkogen.
Diese Mutation ist nicht ungewöhnlich, sie tritt auch schon auf, wenn es Entzündungen oder Schäden in der Bauchspeicheldrüse gibt, die noch nicht bösartig sind. Ab wann aber eine vorgeschädigte Zelle zur Tumorzelle entartet und was genau die gefährliche Veränderung auslöst, ist im Detail bisher nicht bekannt.
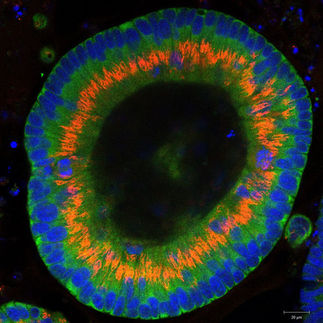
Krebs: Die Zellen entarten nach Entzündungen
Jens Siveke, Gastroenterologe und Spezialist für Krebs im Verdauungstrakt, hat sich zusammen mit einem Team der TU München auf die noch unverstandenen Mechanismen rund um Entzündungen in der Krebsentstehung konzentriert: Welche Zusammenhänge gibt es, welche Akteure und Botenstoffe in der Zelle lösen die bösartige Veränderung aus?
Dabei arbeitete die Gruppe mit den Zellen, die für die Produktion von Verdauungssäften zuständig sind, die sogenannten azinären Zellen. An Mäusen zeigte sich, dass die Entzündung eine Art Auslöser für die bösartige Veränderung ist: Die Entzündungsreaktion der Zellen setzt zahlreiche Botenstoffe frei, die schließlich das bereits mutierte KRAS-Onkogen weiter aktivieren und die Zellentartung fördern.
Die Arbeitsgruppe um Jens Siveke konnte dabei auch weitere Mitspieler im Krebsgeschehen identifizieren, den sog. epidermal growth factor receptor (EGFR), sowie ein Molekül, das eine Schlüsselrolle für wichtige Wachstums- und Teilungsprozesse der Zelle spielt, RAC1 genannt.
In ihren Studien entwickelten sie ein System, mit dem sich das Zusammenspiel von KRAS, EGFR und RAC1 in verschiedenen Erkrankungsstadien studieren ließ: Die Forscher isolierten und kultivierten azinäre Zellen mit den typischen Schädigungen, außerdem untersuchten sie Mäuse bezüglich der Rolle der drei Akteure in vorgeschädigten Zellen einerseits und bereits entarteten Zellen andererseits. Die Mäuse hatten die typische KRAS-Mutation in den Bauchspeicheldrüsenzellen, im Versuch wurde aber der RAC1-Botenstoff bei ihnen mit molekularen Methoden gestoppt.
Entzündungsbotenstoffe unterdrückt
Das überraschende Ergebnis: Fehlte RAC1 durch genetische Manipulation, wurde die Entartung der azinären, vorgeschädigten Zelle zur Tumorzelle komplett unterdrückt. RAC1 ist dabei zuständig für die Aussendung von Entzündungssignalen, die wiederum die Veränderung zur Tumorzelle durch KRAS auslösen.
Zusätzlich konnten die Wissenschaftler zeigen, dass RAC1 hauptsächlich für den äußeren Aufbau der Zelle, ihr Skelett, zuständig ist. Es steuert den Aufbau und die Stabilität dieses Skeletts. Das mutierte Krebsgen KRAS scheint also nur dann auf die Zelle einwirken zu können, wenn auch RAC1 seinen Einfluss auf das Zellskelett ausübt.
Bald Medikamente?
Die Erkenntnis, dass derartige grundlegende Prozesse wie die Regulation des zellulären Aufbaus einen zentralen Einfluss auf die Wirkweise mutierter Onkogene haben, war für Projektleiter Siveke überraschend: „Unsere Versuche zeigen, dass Zellen tatsächlich dauerhaft dem Stress des mutierten Onkogens KRAS widerstehen können. Wir waren sehr erstaunt, dass das überhaupt geht – und es eröffnet Ansätze für ganz neue Therapieverfahren“, sagt Jens Siveke.
Siveke betreut und behandelt viele Patienten mit Pankreaskarzinom und hofft jetzt darauf, dass er, der inzwischen am Westdeutschen Tumorzentrum der Universitätsklinikum Essen und Deutschen Krebsforschungszentrum arbeitet, mit seinem Team auch Medikamente und zielgerichtete Verfahren zur Beeinflussung von RAC1 und KRAS entwickeln kann. „Wir hoffen, mit den besseren Einblicken in die Zell-Mechanismen und im Zusammenspiel mit vielen beteiligten Forschern und Ärzten neuartige Therapiekonzepte zu entwickeln und in klinischen Studien zu testen“.
Die Wilhelm Sander-Stiftung förderte die zweite Phase dieses Forschungsprojekts mit rund 160.000 Euro, nachdem sie die erste Phase mit rund 210.000 € unterstützt hatte.

Weitere News aus dem Ressort Wissenschaft






















































