Wissenschaftler entschlüsseln Defekte in der seltenen Erbkrankheit Myotubuläre Myopathie
Winzige Abweichungen in Körperzellen haben nach einer Studie von Wissenschaftlern aus Berlin, Heidelberg und Strasbourg mitunter schwerwiegende Folgen. Die Gruppe um Prof. Dr. Volker Haucke vom Leibniz-Institut für Molekulare Pharmakologie (FMP) und der Freien Universität Berlin fand heraus, warum Zellen von Patienten, die an der seltenen Muskelerkrankung Myotubuläre Myopathie leiden, nicht richtig funktionieren. Sie ergründeten dabei, wie ein dynamischer, für die Muskelentwicklung und Funktion essenzieller zellulärer Prozess mittels winziger Veränderungen bestimmter Membranlipide gesteuert wird. Die Wissenschaftlerinnen und Wissenschaftler fanden einen Ansatzpunkt für die Entwicklung von Medikamenten, um die schwerwiegende und derzeit unheilbare Erbkrankheit zu behandeln.
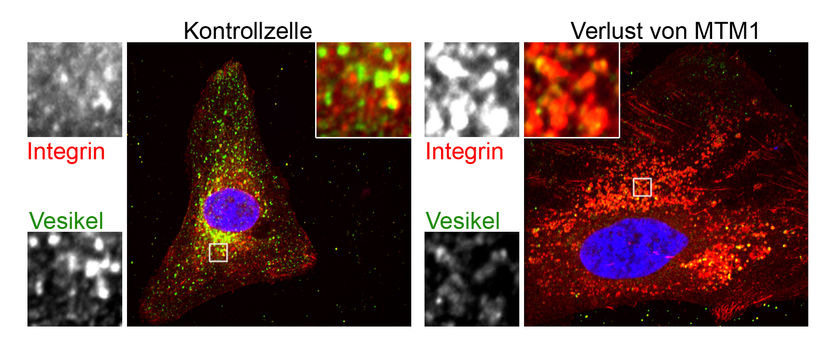
Akkumulation von Integrin (rot), ein wichtiger Baustein von Muskeln, in Vesikeln (grün) aus Zellen ohne MTM1 (rechte Bilder inkl. Vergrößerung) im Vergleich zu Kontrollzellen (linke Bilder inkl. Vergrößerung)
Katharina Ketel, Leibniz Institut für Molekulare Pharmakologie (FMP), Berlin
Kommt ein Kind mit Myotubulärer Myopathie, der schwerwiegendsten Form der zentronukleären Mypopathien (auch als XLCNM bezeichnet) auf die Welt, kann es kaum eigenständig atmen. Die Muskeln sind verkümmert, das Neugeborene liegt schlaff im Arm und ist mitunter sogar zu schwach, um zu trinken. Babys mit der seltenen Muskelerkrankung überleben meist nicht die ersten Lebensmonate. Die Gruppe um Prof. Dr. Volker Haucke hat nun in Zusammenarbeit mit den Laboren von Jocelyn Laporte vom Institut Génétique Biologie Moléculaire Cellulaire (IGBMC) in Strasbourg und Carsten Schultz vom Europäischen Molekularbiologielaboratorium (EMBL) in Heidelberg erforscht, was bei dieser Krankheit auf der molekularen Ebene schiefläuft – und ist dabei auf ein allgemeines Organisationsprinzip in Zellen gestoßen.
Bislang war bekannt gewesen, dass es sich bei der Erbkrankheit um einen Defekt im Gen MTM1 handelt, durch den die Muskelfasern nicht richtig funktionieren. Das Gen liefert den Code für den Bau eines Enzyms, das darauf spezialisiert ist, Phosphatgruppen von den Köpfen bestimmter Membranlipide, den sogenannten Phosphoinositidphosphaten (PIPs) abzuknabbern. Diese PIPs dienen der Zelle zur Markierung ihrer Kompartimente und zur Steuerung des Stofftransports. „Die Zelle ist ein sehr dynamisches System, das wir uns wie eine Metropole vorstellen können, in der sich die Menschen hin- und her bewegen“, erklärt Volker Haucke. „Je nach Anlass ziehen sich die Menschen um – im Frack nimmt man ein Stück weit eine andere Identität an, als wenn man in Jeans und Sweatshirt daherkommt, im Pyjama wird man nicht in die Oper eingelassen. In ähnlicher Weise kleiden sich die Kompartimente und Transportvesikel innerhalb der Zellen in immer wieder andere PIPs und wechseln dadurch ihre Identität.“ Jedes PIP besteht dabei aus einem fettlöslichen Schwanz, der in den Membranen der Zellkompartimente verankert ist und einem wasserlöslichen Kopf, der aus der Membran herausragt. Der Kopf kann an verschiedenen Stellen mit Phosphaten bestückt sein; die Phosphatgruppen werden durch Enzyme abgelöst und an anderen Stellen angefügt. Es handelt sich dabei um eine minimale Veränderung, die blitzschnell vonstattengeht, die aber von der Zelle eindeutig abgelesen werden kann. So ist zum Beispiel durch die Anheftung einer Phosphatgruppe an einer bestimmten Position klar, dass ein Transportbehälter ins Zellinnere gehört; bei einer anderen Bestückung mit Phosphat wandert er zur äußeren Zellmembran, dockt dort an und entlässt seine Fracht ins Freie.
Ein solcher Transport kommt bei den Patienten mit Myotubulärer Myopathie zum Erliegen, wie Katharina Ketel aus der Haucke-Gruppe mit trickreichen Experimenten und hochaufgelösten Aufnahmen aus dem Zellinneren zeigen konnte. Die Ursache der Erkrankung ist ein Gendefekt in MTM1, ein Enzym, das Phosphatgruppen von den Phosphoinositidphosphaten, also den PIPs, entfernt und nur in Kooperation mit einem anderen Enzym arbeitet, das an eine andere Stelle des Kopfes eine Phosphatgruppe anfügt. Es wird dadurch klarer, wodurch die dynamischen Abläufe in Zellen dirigiert werden und illustriert, wie das Studium einer seltenen Erbkrankheit zur Entdeckung molekularer Mechanismen führen kann, die für das Funktionieren unserer Zellen notwendig sind. „Es werden niemals einfach nur beliebige Phosphatgruppen von PIPs entfernt, denn dann stünde ein Zellkompartiment plötzlich ganz ohne Identität da – das käme einem Gedächtnisverlust gleich, es wüsste nicht mehr, woher es kommt und wo es hin soll“, erklärt Volker Haucke. „Durch die Zugabe von synthetischen PIPs mit einem bestimmten Code, konnten wir den Transport der Container manipulieren und damit zeigen, dass die Konversion der PIP-Identität tatsächlich das Problem in Zellen von Patienten mit Myotubulärer Myopathie darstellt“, erläutert Carsten Schultz.
„Bei Patienten mit Myotubulärer Myopathie stranden manche der Transportbehälter im Zellinneren, die eigentlich Proteine zur Zelloberfläche befördern müssten, weil eine Phosphatgruppe bei einem bestimmten PIP nicht entfernt werden kann“, sagt Jocelyn Laporte, ein Experte für diese Krankheit und Koautor der Studie. „In Muskeln gelangen daher Proteine, die für ihre Bildung, Integrität und Funktion notwendig sind, nicht an den richtigen Ort in der Zelle.“ Bei ihren Experimenten in Zellkultur konnten die FMP-Forscher den – krankheitsbedingt unterbundenen – Transport mit einem bestimmten Wirkstoff wieder in Gang setzen. Dies wäre ein Ansatzpunkt für die Entwicklung von Medikamenten, um die schwerwiegende und derzeit unheilbare Erbkrankheit zu behandeln.
Originalveröffentlichung
Weitere News aus dem Ressort Wissenschaft





















Meistgelesene News











Weitere News von unseren anderen Portalen





















Zuletzt betrachtete Inhalte
MoBiTec GmbH - Göttingen, Deutschland
opti-color Meß- und Regelanlagen GmbH - Bad Salzdetfurth, Deutschland
