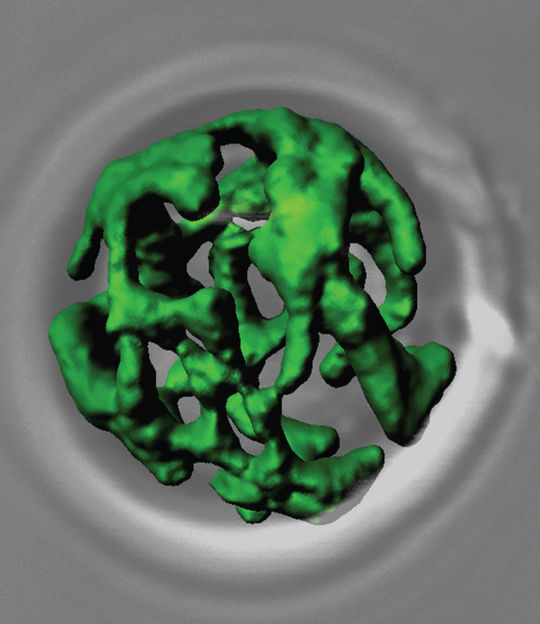
Die Helikase - eine Raupe im Erbgut
Eine Simulation zeigt, wie ein Enzym die RNA-Stränge des Hepatitis-C-Virus trennt
Um wirksame Medikamente gegen Viren zu identifizieren möchten Wissenschaftler verstehen, wie sich diese Krankheitserreger vermehren. Denn um den menschlichen Körper von heimtückischen Viren zu befreien, ist es sehr effektiv, deren Reproduktionsmechanismus außer Kraft zu setzen. Mit ihren Modellrechnungen am Computer sind Forscher vom Fritz-Haber-Institut der Max-Planck-Gesellschaft in Berlin dem Verständnis dieses Mechanismus einen Schritt näher gekommen. Anhand der Helikase des Hepatitis-C-Virus simulierten sie, wie dieses Enzym die Doppelhelixstruktur des viralen Erbguts aufbricht und dieses so für die Reproduktion aufbereitet.
Ohne Helikasen könnten Viren sich nicht vermehren. Sie nutzen diese Enzyme, um ihre eigene RNA oder die DNA der Wirtszelle aufzuspalten - einer der wichtigsten Schritte, um ein neues Virus herzustellen. Nach welchem Muster die Helikase dabei etwa im Hepatitis-C-Virus vorgeht, konnten Wissenschaftler bisher nur erahnen - ihre Struktur ist zu komplex, um sie mit Mikroskopen detailliert zu beobachten. Daher entwickeln Biophysiker Computermodelle, um die komplexen Mechanismen der Virenreproduktion zu verstehen.
Holger Flechsig und Alexander Mikhailov vom Fritz-Haber-Institut der Max-Planck-Gesellschaft haben mit einem neuen Modell simuliert, wie die Helikase des Hepatitis-C-Virus die Helixstruktur seines Erbguts aufspaltet. Für ihre Untersuchung nutzten sie ein Modell aus Hepatitis-C-Helikase und DNA. "Es ist uns gelungen, den vollständigen Arbeitszyklus dieses wichtigen Enzyms mit großer Auflösung zu verfolgen", so Flechsig. "Auch stimmen unsere Ergebnisse prima mit jenen experimenteller Arbeiten überein, die ebenfalls kürzlich veröffentlich wurden." Seine Simulationen bestätigen, was Forscher schon länger vermutet hatten: Das Enzym klettert die DNA Base für Base entlang, indem es sich verformt. Dabei trennt es die beiden Molekülstränge voneinander.
Das Helikaseenzym besteht aus drei Modulen, die ein Dreieck bilden. In dieses Dreieck fädelt sich ein DNA-Strang. Um die Bindungen zwischen den Basenpaaren der DNA-Doppelhelix aufzuspalten, kriecht die Helikase dann wie eine Raupe den DNA-Strang entlang, wie Flechsigs Berechnungen ergaben. Dabei hangeln sich zwei an dem Strang liegenden Module von Base zu Base, indem sie sich einander nähern und sich wieder voneinander entfernen. Währenddessen drückt das dritte Modul, das sich zwischen den beiden DNA-Strängen befindet, gegen die gegenüberliegende Helixhälfte und bricht die Bindungen zu der Partnerbase auf. Wie sich in der Simulation auch zeigte, reichte die Energie eines ATP-Pakets genau, um die Bindung zwischen zwei gegenüberliegenden Basen zu brechen und den Schritt zur nächsten Base zurückzulegen.
Wenn die Forscher die Arbeit von Enzymen am Computer nachahmen, ersetzen sie deren Aminosäuren jeweils durch eine Punktmasse und verbinden sie mit Spiralfedern. Bislang konnten sie durch stark vereinfachte Modelle nur näherungsweise bestimmen, wie sich diese Punktmassen in ihren Bewegungen gegenseitig beeinflussen. Sie berechneten zunächst kleinste Abweichungen der Punktmassen von ihrer lokalen Gleichgewichtsposition. Daraus schlossen sie dann auf die globale Verformung des Enzyms. In einigen Fällen lässt diese sich so auch recht gut beschreiben. Für bestimmte Proteine versagen diese vereinfachten Modelle jedoch. Zudem berücksichtigen sie nicht die Wechselwirkungen zwischen Helikase und DNA.
In ihren neuen Berechnungen beschreiben Flechsig und Mikhailov die Wechselwirkungen zwischen den Punktmassen zwar auch durch mechanische Kräfte. Aber sie modellieren, wie sich die Hepatitis-Helikase global verformt und dies, ohne die mathematische Beschreibung der Bewegungen zu vereinfachen. Darüber hinaus beziehen sie die Kräfte zwischen Helikase und DNA ein. Mit dieser Methode rechneten sie aus, wie die Helikase sich mithilfe der aus dem ATP gewonnenen Energie elastisch verformt und wie sie diese Deformation gezielt für ihre Fortbewegung entlang des Erbguts nutzt.
Inzwischen setzen die Berliner Wissenschaftler ihr Modell auch ein um zu verstehen, wie andere Helikasen funktionieren. Denn viele dieser Enzyme weisen strukturelle Gemeinsamkeiten mit der Hepatitis-C-Helikase auf. Und wenn es gelingt, die Virenvermehrung im Allgemeinen besser zu verstehen, lässt dies auf effektivere Therapien gegen viele Krankheiten hoffen.
Originalveröffentlichung: Holger Flechsig and Alexander S. Mikhailov; "Tracing entire operation cycles of molecular motor hepatitis C virus helicase in structurally resolved dynamical simulations"; PNAS, Advance Online Publication, 16. November 2010
Weitere News aus dem Ressort Wissenschaft





















Meistgelesene News












Weitere News von unseren anderen Portalen






















Zuletzt betrachtete Inhalte

Tests von möglichem Coronavirus-Impfstoff können kommen - US-Minister gibt grünes Licht
Polychlorierte_Dibenzodioxine_und_Dibenzofurane
Die Verlockungen des Immunsystems: Wie sich Abwehrzellen bewegen - Erste Peter Hans Hofschneider Stiftungsprofessur für Molekulare Medizin geht an Dr. Michael Sixt vom Max-Planck-Institut für Biochemie in Martinsried

Hauschild SpeedMixer: Globale Klage gegen das US-Unternehmen FlackTek Inc. und seine europäischen Partner - Hauschild hat eine Klage in den USA, ein Zivilverfahren in den Niederlanden sowie ein Strafverfahren in Spanien gegen FlackTek und seine verschiedenen Vertreter eingeleitet
Henry_Head
Nobelpreisträger und DNA-Mitentdecker Maurice Wilkins gestorben