Falscher Adresscode mit fatalen Folgen: Die zellulären Mechanismen schwerer Neurodegeneratonen
Ein Forscherteam um Professor Christian Haass und Dr. Dorothee Dormann, LMU München und Deutsches Zentrum für neurodegenerative Erkrankungen (DZNE), konnte erstmals zelluläre Mechanismen identifizieren, die zu einer Amyotrophen Lateralsklerose (ALS) oder zu einer Frontotemporalen Demenz (FTD) führen. ALS ist eine schwere Erkrankung des Nervensystems mit zunehmender Muskelschwäche und Lähmungen, während das Absterben von Nervenzellen im Stirnhirn bei Patienten mit FTD Veränderungen der Persönlichkeit sowie Sprach- und Gedächtnisstörungen verursacht.
Eines aber ist beiden Erkrankungen gemein: Proteine, die normalerweise wichtige Aufgaben im Zellkern erfüllen, sammeln sich im Zellkörper an - und führen dort zu krankhaften Ablagerungen. Das Team um Haass konnten zeigen, dass bereits bekannte genetische Mutationen die „Adressaufkleber“ der Proteine gewissermaßen unleserlich machen, so dass die Moleküle am falschen Bestimmungsort ankommen. „Diese Ergebnisse tragen wesentlich dazu bei, die Entstehungsmechanismen der ALS wie auch der Frontotemporalen Demenz besser zu verstehen“, sagt Haas. Nun wollen die Forscher untersuchen, ob die typischen Krankheitssymptome durch die Ablagerungen im Zellkörper oder aber durch den Funktionsverlust der Proteine im Zellkern entstehen.
Bei der Amyotrophen Lateralsklerose (ALS) führt eine irreversible Schädigung der für die Bewegung zuständigen Motoneurone zu einer progressiven und schnell voranschreitenden Muskelschwäche, was Störungen beim Gehen, Sprechen und Schlucken sowie eine Atemlähmung verursacht. Bei der Frontotemporalen Demenz, der nach der Alzheimerschen Erkrankung zweithäufigsten Demenz, bewirkt der massive Abbau von Nervenzellen im Stirnhirn schwere Veränderungen der Persönlichkeit und häufig auch Gedächtnisstörungen. Etwa zehn Prozent der ALS-Patienten leiden unter familiärer ALS, die dominant vererbt und an alle Nachkommen weitergegeben wird. Mutationen des TDP-43-Gens auf Chromosom 1 und dem FUS-Gen auf Chromosom 16 sind bei einer kleinen Zahl der Fälle wichtig. „Diese Proteine scheinen aber bei sehr viel mehr Fällen eine Rolle zu spielen - wie auch bei der Frontotemporalen Demenz“, sagt Professor Christian Haass von der LMU München und vom Deutschen Zentrum für Neurodegenerative Erkrankungen (DZNE).
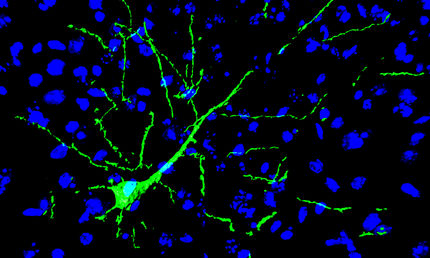
Beim gesunden Menschen befindet sich die Mehrzahl dieser Moleküle im Zellkern der Neurone und hilft dort bei der Übertragung genetischer Information in Proteine. In den Nervenzellen von ALS- oder FTD-Patienten sammeln sich die Proteine aber im Zellkörper und bilden dort krankhafte Ablagerungen. „Bislang war unbekannt, warum derartige Stress-Körnchen entstehen“, so Haass. Gemeinsam mit Wissenschaftlern des Instituts für Neuropathologie in Zürich, dem Leibniz-Institut für Altersforschung in Jena und dem Vancouver General Hospital in Vancouver (Kanada) untersuchte sein Team deshalb, welche zellulären Mechanismen das FUS-Protein bei ALS im Zellkörper akkumulieren lassen. Dabei konnten die Wissenschaftler erstmals nachweisen, dass die bereits bekannten genetischen Defekte den „Adresscode“ von Proteinen betreffen: Bei ALS ist dieses Signal so stark verändert, dass die FUS-Proteine statt in den Zellkern in den Zellkörper und damit an den falschen Bestimmungsort gelangen.
Die Forscher konnten zudem einen Zusammenhang zwischen der Schwere der genetischen Beeinträchtigung und dem Erkrankungsalter sowie dem Schweregrad von ALS beobachten. Eine späte Form beginnt im Alter von über 40 Jahren, während eine frühe Form bereits im Alter von unter 30 Jahren zum Ausbruch kommt. „Uns gelang der Nachweis, dass der genetische ‚Adresszettel‘ bei früh erkrankten Patienten nahezu unlesbar ist“, so Haass. „Ist das Signal noch teilweise lesbar, tritt die Erkrankung spät auf und verläuft auch milder. Wir konnten auch zeigen, dass die fehlgeleiteten Proteine im Zellkörper offenbar dann verklumpen, wenn sie Hitzestress oder Alterungsprozessen ausgesetzt sind.“ Die Ergebnisse lassen die Entstehungsmechanismen von ALS wie auch FTD besser verstehen. Weiterführende Studien sollen nun klären, ob die Symptome durch die Ablagerungen im Zellkörper oder durch den Funktionsverlust der Proteine im Zellkern entstehen.
Originalveröffentlichung: Dorothee Dormann et al.; „ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import”; The EMBO Journal online, 6. Juli 2010
Weitere News aus dem Ressort Wissenschaft




















Diese Produkte könnten Sie interessieren

Antibody Stabilizer von CANDOR Bioscience
Protein- und Antikörperstabilisierung leicht gemacht
Langzeitlagerung ohne Einfrieren – Einfache Anwendung, zuverlässiger Schutz

DynaPro NanoStar II von Wyatt Technology
NanoStar II: DLS und SLS mit Touch-Bedienung
Größe, Partikelkonzentration und mehr für Proteine, Viren und andere Biomoleküle

Meistgelesene News









Weitere News von unseren anderen Portalen






















Zuletzt betrachtete Inhalte
Können Reaktionen in kondensierten Biomaterialien beschleunigt werden? - Forschungsprojekt “RNA Epicatalysis” wird mit “Experiment!”-Stipendium der Volkswagen Stiftung gefördert
Musculus_gastrocnemius
Roquin erkennt neu entdecktes RNA-Motiv in Genen

Klebstoffe aus Restholz, Biosprit aus Stroh - EU-Forschungsprojekt zu den Potenzialen von Agrar- und Forstabfällen als umweltfreundliche Rohstoffe abgeschlossen
Selbsttoleranz

Handynutzung erhöht nicht das Risiko von Hirntumoren

Eine echte Alternative zum Erdöl - Synthese biobasierter Hochleistungs-Polyamide aus biogenen Reststoffen
