Blutarmut mit Genschere behandeln
Gentherapien, die häufig vorkommende genetisch bedingte Blutarmutserkrankungen beheben könnten

Viele Erbkrankheiten gelten bislang als unheilbar. Zu unberechenbar und kompliziert der Eingriff ins Erbgut, zu ungewiss der Ausgang der Veränderung. Denn oft ist nicht nur ein Gen ins Krankheitsgeschehen involviert, sondern mehrere, die auf verschiedenen Chromosomen liegen können.
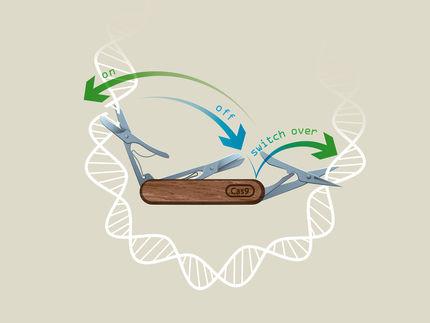
Seit sich die Nutzung der Genschere CRISPR/Cas9 fast schon explosionsartig verbreitet hat, haben sich die Spielregeln massiv verändert. Die gezielte Veränderung von einzelnen Genen oder sogar Bausteinen des Erbguts haben dadurch innerhalb der vergangenen Jahre einen grossen Sprung nach vorne gemacht. Dank dieses gemeinsamen Efforts von Forschenden weltweit rückt nun die Heilung von Erbkrankheiten beim Menschen in Griffweite.
Genschere gegen beta-Hämoglobinopathien einsetzen
Einer Erbkrankheit mit der CRISPR/Cas9-Technik zu Leibe rücken will auch die Molekularbiologin Mandy Boontanrart aus der Gruppe von ETH-Professor Jacob Corn. Soeben hat sie an einer Studie mitgearbeitet, die sich als bahnbrechend für die Behandlung von vererbbaren beta-Hämoglobinopathien – zwei Formen von Blutarmut – erweisen könnte. Dazu zählen die beta-Thalassämie und die Sichelzellanämie, zwei der weltweit häufigsten Erbkrankheiten.

Verursacht werden beta-Hämoglobinopathien durch Mutationen des HBB-Gens. Dieses ist der Bauplan für eine Proteinkette namens beta-Globin, ein Bestandteil des roten Blutfarbstoffs Hämoglobin, der millionenfach in roten Blutkörperchen steckt und für den Sauerstofftransport im Körper zuständig ist. In der Regel ist Hämoglobin bei Erwachsenen aus zwei alpha-Globinen und zwei beta-Globinen zusammengesetzt. In geringem Umfang liegt auch Hämoglobin vor, das aus zwei alpha- und zwei delta-Globinen besteht. Letzteres funktioniert gleich wie beta-Globin, wird aber in roten Blutzellen natürlicherweise nur in sehr kleinen Mengen hergestellt.
Wenn nun eine Mutation des HBB-Gens dazu führt, dass beta-Globine fehlerhaft hergestellt werden, kommt es zu einem Mangel an funktionierendem Hämoglobin. Typischerweise kann dies dazu führen, dass rote Blutzellen frühzeitig absterben. Es kommt zur Blutarmut. Die Organe und der gesamte Körper sind dann chronisch mit Sauerstoff unterversorgt.
Ist nur eine Kopie des HBB-Gens mutiert, können Träger:innen der Mutation ein mehr oder weniger normales Leben führen. «Zwar wird eine Person mit einer Mutation Schwierigkeiten haben, Profi-Athletin zu werden, aber man kann trotzdem Joggen, Schwimmen und Radfahren», sagt Boontanrart, die selbst Trägerin eines mutierten Gens ist. Sind beide Genkopien schadhaft, wird es problematisch: «Möchte man Kinder haben mit einem Partner, der die Mutation auch aufweist, könnten die Kinder beide mutierten Gene erhalten, eines vom Vater, eines von der Mutter. Diese Kinder wären schwer krank.»
delta-Globin-Produktion ankurbeln
Eine wirksame Behandlung für beta-Hämoglobinopathien ist jedoch nicht verfügbar. In ihrer neuen Studie zeigen Boontanrart und ihre Kolleg:innen, dass sich das Problem über die Förderung der delta-Globin-Bildung, welches das schadhafte beta-Globin ersetzt, lösen liesse. «Der Mensch produziert von Natur aus nur geringe Mengen von delta-Globinen. Das hat mit einer speziellen DNA-Steuerungssequenz zu tun, die die Transkription des entsprechenden Gens verhindert.» Die Forschenden kamen deshalb auf die Idee, diese Steuerungssequenz so zu verändern, dass die delta-Globin-Produktion angekurbelt wird.
Um dies zu bewerkstelligen, verwendete Boontanrart die CRISPR/Cas9-Genschere. Damit fügte sie in Blutvorläuferzellen vor dem HBD-Gen, den Bauplan von delta-Globin, drei zusätzliche DNA-Abschnitte ein. Diese regen die Zellmaschinerie dazu an, mehr delta-Globin zu erzeugen – was tatsächlich gelungen ist.
Die Resultate sind vielversprechend: «Wir konnten den delta-Globin-Anteil markant steigern, und zwar auf ein Niveau, das therapeutisch wirksam sein könnte», sagt die Forscherin.
Das Einfügen mehrerer DNA-Elemente sei allerdings noch immer eine grosse Herausforderung. «Es ist anspruchsvoller als die Technik, die andere Forschungsgruppen und Pharmafirmen anwenden», betont Boontanrart. Forschende in den USA sind ebenfalls daran mithilfe des CRISPR/Cas9-Systems beta-Hämoglobinopathien anzugehen, indem sie Blutstammzellen dazu bringen, fetales Hämoglobin zu produzieren. Dieses ist die vorherrschende Hämoglobin-Form in Föten. Die Produktion stoppt spätestens einige Monate nach der Geburt. Fetales Hb soll bei der neuen Therapie anstelle des beta-Globins treten. Dieser Ansatz wird derzeit von der Federal Drug Administration (FDA) für die Zulassung geprüft.
Obschon weit gediehen, habe dieses Vorgehen aber Tücken, sagt Boontanrart. Es lasse sich beispielsweise nicht bei Frauen anwenden, die schwanger sind oder werden möchten, weil fetales Hämoglobin Sauerstoff stärker bindet als Hämoglobin von Erwachsenen. Dadurch könnte die Mutter ihrem ungeborenen Kind den Sauerstoff wegschnappen.
«Die delta-Globin-Produktion zu steigern, ist das meiner Meinung nach die bessere therapeutische Option. Delta-Hämoglobin hat sehr ähnliche Eigenschaften wie beta-Globin und kann für nahezu alle Patienten verwendet werden», sagt Boontanrart.
Spin-off ist im Aufbau
Um ihre Forschungsresultate in die Praxis zu übertragen, begann Boontanrart 2021 während ihres ETH Pioneer Fellowships das Ariya Bio-Projekt. Den Sitz hat es im ieLab der ETH in Schlieren vor den Toren der Stadt Zürich. 2022 hat die ETH Zürich zudem einen Patentantrag gestellt, um die Entwicklung zu schützen.
Jetzt ist Boontanrart daran, mit zwei Anschluss-Grants, einem SNSF Bridge Fellowship und Mitteln von Innosuisse, der Innovationsförderung des Bundes, präklinische Untersuchungen vorzubereiten. Diese Untersuchungen sollen im September starten. Mit diesen Studien wollen die Forschenden den Therapieansatz erstmals an Tieren testen, um herauszufinden, ob er sicher und in lebenden Organismen wirksam ist. Bisherige Versuche wurden in Zellkultur durchgeführt.
Die Forscherin hofft, dass bis 2030 alle klinischen Tests abgeschlossen sein werden und ein Produkt verfügbar sein wird. «Ich bin optimistisch, dass wir die Zulassung schneller erhalten als die Geneditierungsverfahren, die heute in Überprüfung sind, denn sie helfen mit, unserem Ansatz den Weg zu ebnen.»
Originalveröffentlichung
Weitere News aus dem Ressort Wissenschaft




















Meistgelesene News










Weitere News von unseren anderen Portalen






















Da tut sich was in der Life-Science-Branche …
So sieht echter Pioniergeist aus: Jede Menge innovative Start-ups bringen frische Ideen, Herzblut und Unternehmergeist auf, um die Welt von morgen zum Positiven zu verändern. Tauchen Sie ein in die Welt dieser Jungunternehmen und nutzen Sie die Möglichkeit zur Kontaktaufnahme mit den Gründern.