Proteintröpfchen als Ursache vieler genetischer Krankheiten
Fehlfunktion von zellulären Kondensaten könnte angeborenen Fehlbildungen, Volkskrankheiten und Krebs zugrunde liegen
Die meisten Proteine sammeln sich in proteinreichen Tröpfchen in der Zelle, den „zellulären Kondensaten“. Solche Proteine besitzen Abschnitte, die als Adressetikett dienen: Sie zeigen das Kondensat an, für das das Protein bestimmt ist. Wenn die Etiketten fehlerhaft sind, können die Proteine im falschen Kondensat landen. Dies könnte laut einem Team von Forschenden aus der klinischen Medizin und der biologischen Grundlagenforschung die Ursache zahlreicher unaufgeklärter Erkrankungen sein. Ihre Ergebnisse sind im Fachjournal Nature erschienen.
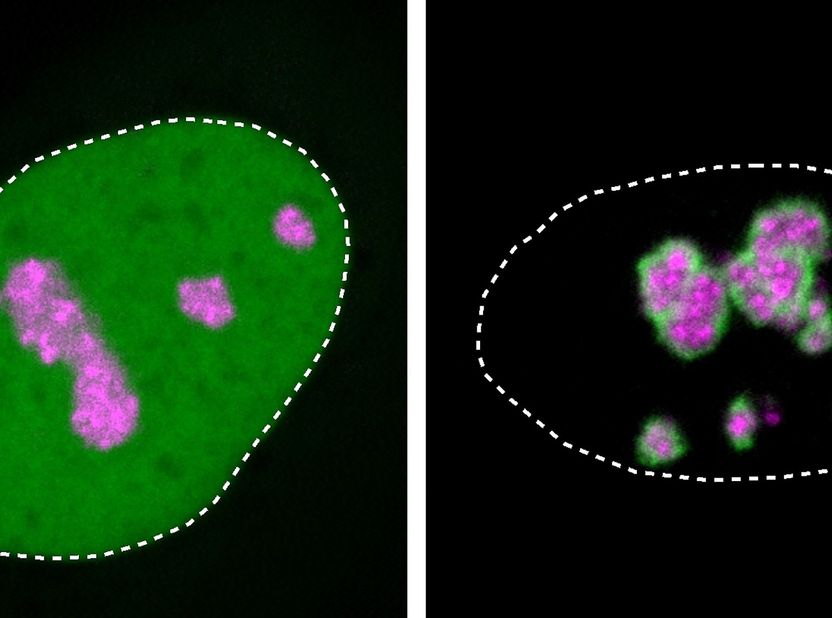
Detail von Zellkernen in humanen Zellkulturen: Das HMGB1-Protein (grün) ist normalerweise im Zellkern verteilt (gepunktete Linie). Rechts ist die Mutation von HMGB1 zu sehen, die sich am Kernkörperchen (pink) eine harte Schicht aufbaut und so die Krankheit verursacht.
Henri Niskanen, MPIMG
Betroffene mit BPTA-Syndrom besitzen charakteristisch fehlgebildete Gliedmaßen mit kurzen Fingern und überzähligen Zehen, fehlenden Schienbeinen und ein verkleinertes Gehirn. Wie die Forschenden herausfanden, ist der Grund dafür eine besondere genetische Veränderung in einem wichtigen Protein. Diese lässt das Eiweiß in das Kernkörperchen wandern, einem proteinreichen Tröpfchen im Zellkern. Die Folge: Die Funktion des Kernkörperchens ist beeinträchtigt und es kommt zur Entwicklungsstörung.
„Was wir bei dieser einen Krankheit entdeckt haben, scheint für viele weitere Krankheiten zu gelten. Es ist also nicht das seltene Einhorn, das es nur einmal gibt. Vielmehr konnten wir das Phänomen bislang nur nicht sehen, weil wir nicht wussten, wie wir danach suchen sollten“, sagt Denise Horn, klinische Genetikerin vom Institut für Medizinische Genetik und Humangenetik der Charité – Universitätsmedizin Berlin.
Zusammen mit Wissenschaftlerinnen und Wissenschaftlern vom Max-Planck-Institut für Molekulare Genetik (MPIMG) in Berlin, vom Universitätsklinikum Schleswig-Holstein (UKSH) in Kiel und Lübeck und Mitwirkenden aus der ganzen Welt stoßen die Forschenden damit eine Tür für neue Diagnosen auf, die zur Aufklärung zahlreicher weiterer Erkrankungen führen könnte sowie zu möglichen zukünftigen Therapien.
„Wir haben einen neuen Mechanismus entdeckt, der bei einer großen Bandbreite von Krankheiten im Spiel sein könnte, darunter Erbkrankheiten und Krebs“, sagt Denes Hnisz, Forschungsgruppenleiter am MPIMG. „Tatsächlich haben wir ähnliche Veränderungen in mehr als 600 weiteren Proteinen entdeckt, von denen 101 bekannterweise mit Krankheiten in Verbindung stehen.“
„Die eigentliche Arbeit beginnt erst jetzt“, fügt Malte Spielmann, Direktor des Instituts für Humangenetik des UKSH, hinzu. „Wir werden viele weitere Gene mit krankheitsauslösenden Mutationen finden und können nun ihren Mechanismus aufklären.“
Eine außergewöhnliche Mutation
Die Betroffenen haben auffällige Fehlbildungen an den Gliedmaßen, im Gesicht und im Nerven- und Knochensystem, die durch die komplizierte Krankheitsbezeichnung „Brachyphalangie-Polydactylie und tibiale Aplasie/Hypoplasie-Syndrom“ (BPTAS) nur teilweise beschrieben sind.
„Mit weniger als zehn dokumentierten Fällen weltweit ist die Krankheit nicht nur selten, sondern extrem selten“, sagt Martin Mensah vom Institut für Medizinische Genetik und Humangenetik der Charité. Um der Ursache auf die Spur zu kommen, entschlüsselte er zusammen mit den Kolleginnen und Kollegen das Genom von fünf Betroffenen und stellte fest, dass in allen das Gen für das Protein HMGB1 verändert war.
Dieses Protein hat die wichtige Aufgabe, das Erbgut im Zellkern zu organisieren und vermittelt die Interaktion von Molekülen mit der DNA, etwa um Gene abzulesen.
Bei Mäusen ist ein vollständiger Verlust des Gens auf beiden Chromosomen verheerend und führt zum Tod des Embryos. Bei Patienten, bei denen nur eine Kopie mutiert ist, nutzen die Zellen jedoch die intakte Kopie auf dem anderen Chromosom, was nur zu einer leichten neurologischen Entwicklungsverzögerung führt. Aber die neu entdeckten Fälle passen nicht in dieses Schema.
„Die fünf Personen, die alle nicht miteinander verwandt waren, wiesen die gleiche extrem seltene Erkrankung auf und hatten im Grunde dieselbe Mutation“, sagt Mensah, der auch Fellow des Clinician Scientist Programms des Berlin Institute of Health in der Charité (BIH) ist. „Deswegen sind wir sicher, dass die HMGB1-Mutation die Ursache für die Krankheit ist. Allerdings hatten wir damals noch keine Ahnung, wie das Genprodukt die Krankheit funktionell verursacht, zumal Funktionsverlust-Mutationen Berichten zufolge ganz andere Folgen haben.“
Geladene Proteinfortsätze
Bei genauerem Hinsehen stellte sich heraus, dass verschiedene Mutationen von HMGB1 unterschiedliche Effekte haben. Wie die Sequenzierungsdaten zeigten, ist in den Betroffenen mit den schweren Fehlbildungen das Leseraster für das letzte Drittel des HMGB1-Gens verschoben.
Nach der Übersetzung in ein Protein ist der korrespondierende Bereich nun nicht länger mit negativen, sondern mit positiv geladenen Aminosäure-Bausteinen ausgestattet. So etwas kann passieren, wenn Vielfache von drei genetischen Buchstaben in der Sequenz fehlen. Denn jeweils drei aufeinander folgende Buchstaben kodieren immer für einen Baustein im Protein.
Der hintere Teil des Proteins besitzt jedoch keine definierte Struktur. Stattdessen hängt dieser Abschnitt wie ein loses Gummiband aus dem Molekül heraus. Die Aufgaben solcher Proteinschwänze (auch „intrinsisch ungeordnete Regionen“ genannt) sind schwer zu erforschen, weil sie häufig erst zusammen mit anderen Molekülen ihre Wirkung entfalten. Wie also könnte seine Mutation zur beobachteten Krankheit führen?
Proteintröpfchen in der Zelle
Um diese Frage zu beantworten, traten die Medizinerinnen und Mediziner mit ihren Patientenproben an den Biochemiker Denes Hnisz und Henri Niskanen am MPIMG heran, die sich in ihrer Arbeit mit zellulären Kondensaten befassen, die wichtige Gene steuern. Diese tropfenartigen Gebilde verhalten sich etwa so wie die Öl- und Essigtropfen in einem Salatdressing. Sie bestehen aus einer Vielzahl unterschiedlicher Moleküle, sind von ihrer Umgebung abgegrenzt und können sich dynamisch verändern.
„Wir denken, dass Kondensate sich in der Zelle aus praktischen Gründen bilden“, erklärt Niskanen. Moleküle für eine bestimmte Aufgabe würden so zusammengefasst, etwa um ein Gen abzulesen. Allein dafür seien mehrere hundert Proteine notwendig, die es an den richtigen Ort zu schaffen gälte.
„Die intrinsisch ungeordneten Regionen, die häufig keine offensichtliche biochemische Funktion haben, sind vermutlich für die Bildung von Kondensaten verantwortlich“, sagt Niskanen und beschreibt anhand eines Beispiels, wie wichtig die physikalischen Eigenschaften der Proteinfortsätze dafür sind. „Aus vielen losen Gummibändern lässt sich leicht ein Ball herstellen, der relativ fest zusammenhängt und mit wenig Mühe wieder auseinanderzunehmen ist. Ein Knäuel aus glatter Angelschnur oder aus Klebeband würde sich dagegen ganz anders verhalten.“
Tröpfchen verfestigen sich
Auch das Kernkörperchen im Zellkern ist ein solches zelluläres Kondensat, das unter dem Mikroskop wie ein diffuser dunkler Fleck aussieht. Dort halten sich vorzugsweise Moleküle mit positiv geladenen Schwänzen auf. Viele davon sind an der Proteinsynthese beteiligt und sind damit essentiell für die Zelle.
Das mutierte Protein HMGB1 mit seinem nun positiv geladenen Molekülschwanz wird ebenfalls zum Kernkörperchen hingezogen, wie das Team anhand von Versuchen mit isolierten Proteinen und mit Zellkulturen beobachten konnte.
Weil die mutierte Proteinregion auch einen ölig-klebrigen Teil hinzugewonnen hat, verklumpt es außerdem. Das Kernkörperchen verliert seine flüssigkeitsähnlichen Eigenschaften und erstarrt zunehmend, was Niskanen unter dem Mikroskop beobachten konnte. Dies beeinträchtigte auch die Lebensfunktionen der Zellen: Mit dem mutierten Protein starben mehr Zellen in der Kultur, als in einer Zellkultur ohne die Mutation.
Datenbanken durchkämmt
Anschließend durchsuchte das Forschungsteam in Datenbanken Genomdaten von tausenden Personen auf der Suche nach ähnlichen Fällen. Tatsächlich konnten die Forscherinnen und Forscher mehr als sechshundert Mutationen in 66 Proteinen identifizieren, in denen das Leseraster durch eine Mutation im Proteinschwanz so verschoben war, dass dieser sowohl positiv geladen als auch „öliger“ wurde. 101 davon waren bereits zuvor mit verschiedenen Krankheiten in Verbindung gebracht worden.
Für einen Versuch wählte das Team 13 mutierte Gene aus und testete sie in der Zellkultur. In 12 Fällen hatten die mutierten Proteine eine Anziehung für das Kernkörperchen erworben. Etwa die Hälfte der getesteten Proteine beeinträchtigte die Funktion des Kernkörperchens und ähnelte damit dem Krankheitsmechanismus des BPTA-Syndroms.
Neue Erklärungen für existierende Krankheiten
„Für die klinische Forschung könnte unsere Studie einen augenöffnenden Effekt haben“, sagt Malte Spielmann, der die Untersuchungen zusammen mit Denes Hnisz und Denise Horn leitete. „Zukünftig können wir sicher die Ursachen einiger genetisch bedingter Krankheiten aufklären und vielleicht einmal therapieren.“
„Allerdings sind angeborene genetisch bedingte Krankheiten wie das beschriebene BPTAS auch mit dem neuen Wissen kaum zu heilen,“ sagt Horn. „Weil sich die Fehlbildungen bereits im Mutterleib entwickeln, müssen sie dort medikamentös behandelt werden, noch bevor sie entstehen. Dies wäre nur sehr schwer möglich.“
Doch auch Tumorleiden seien auf genetische Veränderungen in den betroffenen Zellen zurückzuführen, fügt Hnisz hinzu: „Zelluläre Kondensate und die damit verbundene Phasenseparation sind ein grundlegender Mechanismus der Zelle, der auch bei Krebs eine Rolle spielt. Die Chancen, hierfür gezielte Therapien zu entwickeln, sind deutlich besser.“
Originalveröffentlichung

Weitere News aus dem Ressort Wissenschaft




















Diese Produkte könnten Sie interessieren

Antibody Stabilizer von CANDOR Bioscience
Protein- und Antikörperstabilisierung leicht gemacht
Langzeitlagerung ohne Einfrieren – Einfache Anwendung, zuverlässiger Schutz

DynaPro NanoStar II von Wyatt Technology
NanoStar II: DLS und SLS mit Touch-Bedienung
Größe, Partikelkonzentration und mehr für Proteine, Viren und andere Biomoleküle

Meistgelesene News








Weitere News von unseren anderen Portalen






















Zuletzt betrachtete Inhalte
Wie Kälte Fruchtbarkeit und Lebensspanne erhöht
Wallenberg-Syndrom

Neue Biomarker für Kaffeekonsum
Guillain-Barré-Syndrom

Von Seegurken lernen: umweltfreundliche Biolacke aus dem Meer? - Oldenburger Forscher haben untersucht, welche Substanzen aus Seegurken Antifouling-Effekte haben

Neandertalergene in der Petrischale - Forscher untersuchen die Effekte der Neandertaler-DNA in heutigen Menschen mit Hilfe von Stammzellen und Organoiden
Anders als bisher gedacht: Die Dynamik von Pluripotenz-Transkriptionsfaktoren
Paris bereitet sich auf die CPhI Worldwide vor - Für die Pharmamesse werden mehr als 25.000 Besucher erwartet

Übermässiges Trinken: Gewohnheit oder Hormonstörung? - Welcher Test liefert eine zuverlässige Diagnose?
Sartorius eröffnet neues Werk in Indien
