Schicksal von Zellen vorhersagen
KI-Lösungen für medizinische Spitzenforschung entwickelt
Daten sind Gold – das gilt nicht nur für die Wirtschaft, sondern auch für die biomedizinische Forschung. Um neue Therapien oder Präventionsstrategien für Krankheiten zu entwickeln, brauchen Wissenschaftler:innen immer schneller immer mehr und immer bessere Daten. Doch die Qualität ist häufig sehr unterschiedlich und die Integration verschiedener Datensätze oft fast unmöglich. Mit dem Computational Health Center des Forschungszentrums Helmholtz Munich entsteht jetzt unter der Leitung von Fabian Theis eines der europaweit größten Forschungszentren für künstliche Intelligenz in der medizinischen Wissenschaft. In enger Vernetzung mit der Technischen Universität München (TUM) entdecken hier mehr als hundert Wissenschaftler:innen mithilfe von künstlicher Intelligenz und maschinellem Lernen Lösungen für genau diese Probleme und ermöglichen damit medizinische Innovationen für eine gesündere Gesellschaft. In der jüngsten Ausgabe des Fachjournals Nature Methods präsentieren sie gleich drei Artikel mit bahnbrechenden neuen Lösungen.
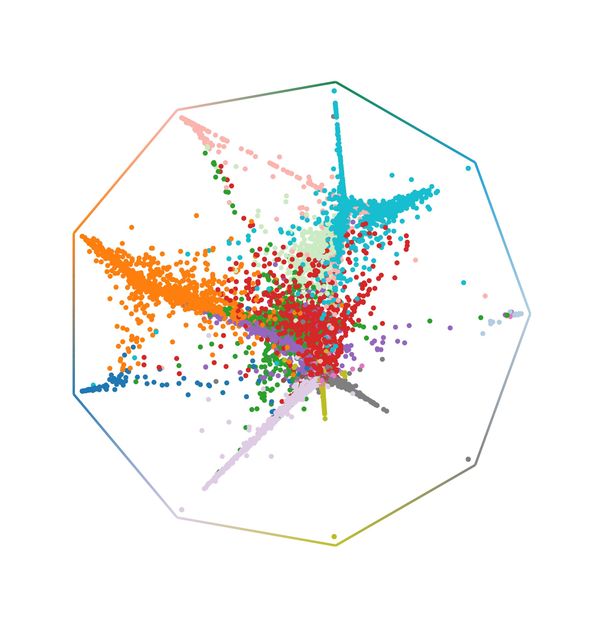
CellRank-Wahrscheinlichkeiten für die Regeneration der Lunge; jede Zelle ist an einer Position platziert, die ihre Wahrscheinlichkeit widerspiegelt, einen beliebigen Endzustand zu erreichen.
© Helmholtz Munich / Marius Lange
Fabian Theis, Leiter des Computational Health Center bei Helmholtz Munich und Professor für Mathematische Modellierung biologischer Systeme an der TUM: „Wir haben vier intensive Wochen hinter uns, in denen unsere wissenschaftlichen Projekte und Methoden gleichzeitig erfolgreich publiziert wurden. In meiner Gruppe konzentrieren wir uns auf die Einzelzellgenomik. Mit dieser Methode wollen wir den Ursprung von Krankheiten auf mechanistische Weise verstehen. Dafür nutzen und entwickeln wir Ansätze des maschinellen Lernens, um komplexe Daten besser darzustellen. Mit unseren drei neuesten Studien haben wir uns mit der Integration von Einzelzelldaten, dem Lernen von Trajektorien und der räumlichen Auflösung beschäftigt. Mit diesen Beiträgen aber auch darüber wollen wir die Einzelzellforschung und damit unser Verständnis von Krankheiten auf die nächste Stufe bringen.“
Die neuesten Lösungen von Helmholtz Munich und der TUM im Überblick:
Das Dilemma mit der Datenintegration lösen
In wissenschaftlichen Studien arbeiten Forschende oft an einzelnen Datensätzen. Um zu prüfen, ob sie ihre Ergebnisse aus einer Studie verallgemeinern können, müssen sie ihre Daten mit anderen Datensätzen aus demselben System vergleichen. In der medizinischen Forschung handelt es sich dabei oft um Daten einzelner Zellen. Da Einzelzelldaten nicht immer zur gleichen Zeit, am gleichen Ort oder von der gleichen Person erzeugt wurden, unterscheiden sich auch die gleichen Zelltypen in ihrem molekularen Profil. Dieses Problem bezeichnet man als Batch-Effekt und es erschwert die Kombination von Datensätzen immens. Bisher brachte die Forschung mehr als fünfzig unterschiedliche Lösungsvorschläge hervor, doch welcher ist der beste? Malte Lücken und seine Kolleg:innen haben 86 Datensätze sorgfältig aufbereitet und 16 der gängigsten Lösungen für die Datenintegration anhand von 13 Aufgaben miteinander verglichen. Nach mehr als 55.000 Stunden Rechenzeit und einer detaillierten Auswertung von 590 Ergebnissen haben sie einen Leitfaden erstellt, wie sich das Dilemma mit der Datenintegration am besten lösen lässt. Dies ermöglicht eine bessere Beobachtung von Krankheitsprozessen über unterschiedliche Datensätze hinweg.
Schicksal von Zellen mit Open-Source-Software vorhersagen
In der medizinischen Forschung dreht sich vieles um die Fragen: Wie entwickeln sich Zellen? Wie funktioniert Zellregeneration? Um diese zu beantworten interessieren sich Forschende für die Genexpression von Zellen, die über eine Methode namens Einzelzell-RNA-Sequenzierung ermittelt wird. Das Verfahren zerstört jedoch die Zelle und liefert nur eine kurze Momentaufnahme der Genexpression. Deshalb haben Wissenschaftler:innen bereits viele Algorithmen entwickeln, um von der Momentaufnahme künstlich auf einen kontinuierlichen Entwicklungsprozess rückschließen zu können. Die Algorithmen stehen jedoch alle vor derselben Herausforderung: Sie können keine verlässlichen Vorhersagen für das Schicksal der Zelle treffen. Marius Lange und seine Kolleg:innen arbeiten dafür an einem neuen Algorithmus. CellRank beschreibt die Entwicklung einer Zelle, indem es die Momentaufnahme der Genexpression mit “RNA Velocity” kombiniert, einem Konzept zur Abschätzung der Genregulation. Sowohl in vitro als auch in vivo konnte CellRank das Schicksal von Zellen korrekt vorhersagen und bekannte Gene wiedererkennen. In einem Beispiel zur Lungenregeneration sagte CellRank neuartige Zellzwischenzustände voraus, deren Existenz experimentell bestätigt wurde. CellRank ist eine Open-Source-Software, die in Laboren weltweit bereits zum Einsatz kommt um komplexe Zelldynamiken im Kontext von Reprogrammierung, Regeneration oder Krebs zu untersuchen.
Räumliche Omics-Analysen visualisieren
In den letzten Jahren wurden immer mehr Technologien entwickelt, um Veränderungen in der Genexpression von Geweben zu messen. Der Vorteil solcher Technologien besteht darin, dass Forschende die Zellen in ihrem Kontext beobachten können. So können sie besser verstehen, wie das Gewebe aufgebaut ist und wie die Zellen miteinander kommunizieren. Um die wachsende Vielfalt solcher Daten speichern, integrieren und visualisieren zu können, braucht es flexible computergestützte Systeme. Zu diesem Zweck haben Giovanni Palla, Hannah Spitzer und Kolleg:innen eine neue Software, Squidpy genannt, entwickelt. Squidpy ermöglicht die Verarbeitung räumlicher Genexpressionsdaten. Es vereint Werkzeuge für die Genexpressions- und die Bildanalyse, um räumliche Omics-Daten effizient zu bearbeiten und interaktiv zu visualisieren. Squidpy ist erweiterbar und kann mit einer Vielzahl von Tools für maschinelles Lernen aus dem Python-Ökosystem verbunden werden. Wissenschaftler:innen auf der ganzen Welt nutzen die Lösung bereits, um räumliche molekulare Daten zu analysieren.
Originalveröffentlichung
Weitere News aus dem Ressort Wissenschaft




















Diese Produkte könnten Sie interessieren

Limsophy LIMS von AAC Infotray
Optimieren Sie Ihre Laborprozesse mit Limsophy LIMS
Nahtlose Integration und Prozessoptimierung in der Labordatenverwaltung
GUS-OS Suite von GUS
Ganzheitliche ERP-Lösung für Unternehmen der Prozessindustrie
Integrieren Sie alle Abteilungen für nahtlose Zusammenarbeit

Meistgelesene News










Weitere News von unseren anderen Portalen






















Zuletzt betrachtete Inhalte
Autoantikörper bei COVID-19 weniger schädlich als gedacht? - Vermutlich trifft sie in den meisten Fällen keine Schuld, wenn es den Patienten besonders schlecht ergeht