Verunglückte Zellteilung treibt Krebszellen kindlicher Tumoren in den Tod
Krebsforscher haben einen Mechanismus entdeckt, mit dem sich die Selbstzerstörung von Krebszellen kindlicher Tumoren aktivieren lässt
Eine Medikamentenkombination, die bereits bei anderen Krebserkrankungen erprobt wird, führte bei Mäusen dazu, dass die Krebszellen Fehler in ihrem Erbmaterial anhäuften und die Knochen- und Weichteiltumoren im Labor schrumpften. Die Wissenschaftler des Hopp-Kindertumorzentrums Heidelberg (KiTZ), des Universitätsklinikums Heidelberg (UKHD) und des Deutschen Krebsforschungszentrums (DKFZ) zeigten zudem, dass ein Schlüsselmolekül der Zellteilung als neuer Biomarker genutzt werden könnte, um die Patienten auszuwählen, die von dieser Therapie profitieren.
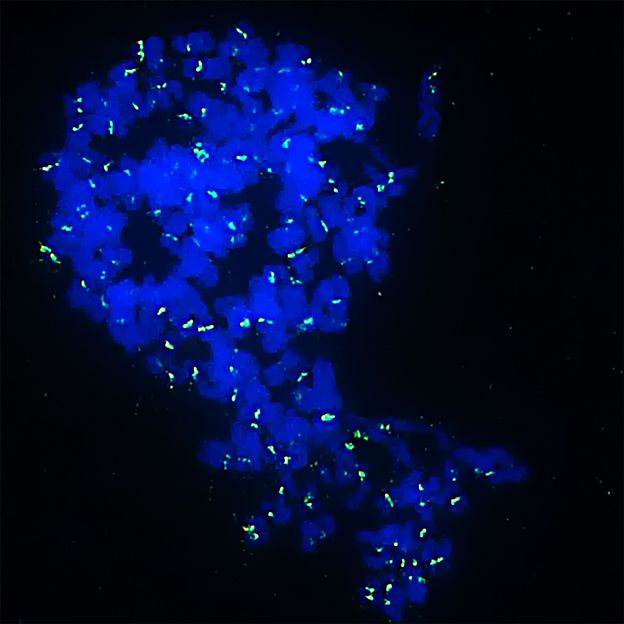
Hemmstoffe bewirken, dass Ewing-Sarkomzellen zu Monsterzellen mit vielfachen Chromosomensätzen (in blau) werden und dann absterben. In Grün sieht man die Stelle der Chromosomen, die beide Chromosomenhälften zusammenhält.
Jing Li/KiTZ
Normalerweise werden genetisch entartete Zellen von einem zellulären Programm in den Selbstmord getrieben und beugen durch ihr „freiwilliges“ Ableben der Krebsentstehung vor. Krebszellen schaffen es jedoch, dem sogenannten programmierten Zelltod, auch Apoptose genannt, zu entgehen und sich weiter zu teilen. Ihr instabiles Erbgut bringt ihnen dabei sogar Wachstumsvorteile und macht sie therapieresistent.
Im Gegensatz dazu verhält sich das Genom bei bestimmten Krebsarten im Kindesalter, wie z. B. dem Ewing-Sarkom, bemerkenswert ruhig, und man findet nur wenige solcher genomischen und chromosomalen Umlagerungen. Ewing-Sarkome sind hochaggressive Tumoren, die sich in Knochen- oder Weichteilgeweben bilden können und hauptsächlich bei Kindern und Jugendlichen vorkommen. „Viele Standardtherapien versagen gerade bei dieser Krebsart, so dass neue Behandlungsoptionen dringend erforderlich sind“, sagt Thomas Grünewald, Leiter der Arbeitsgruppe Translationale Pädiatrische Sarkomforschung am KiTZ und der gleichnamigen Abteilung am DKFZ.
Gemeinsam mit Wissenschaftlern der Ludwig-Maximilians-Universität (LMU) München und des Deutschen Konsortiums für Translationale Krebsforschung (DKTK) suchte das Team um Grünewald einen Weg, sich die genetische Stabilität der kindlichen Krebszellen zunutze zu machen und sie gezielt in den Tod zu treiben. „Die Krebszellen von Kindern reagieren noch auf die Signale des programmierten Zelltods. Unsere Hoffnung war daher, dass wir nur genügend Durcheinander im Genom der Zellen anrichten müssen, damit sie ihre Selbstzerstörung einleiten“, erläutert Grünewald.
Eine geeignete molekulare Schwachstelle hatten die Wissenschaftler schon bei früheren Arbeiten ins Auge gefasst: Das Protein PRC1. „PRC1 trägt zur stabilen Teilung der Tumorzellen bei und wird durch ein mutiertes Krebsgen im Ewing-Sarkom verstärkt gebildet“, erklärt die Erstautorin der Studie Jing Li, die das molekulare Zusammenspiel des mutierten Gens mit PRC1 intensiv untersucht hat.
Durch Kombination zweier Hemmstoffe, die bereits in klinischen Studien gegen andere Krebserkrankungen erprobt werden, gelang es den Wissenschaftlern, die PRC1-Aktivität und damit die Zellteilung der Krebszellen gezielt zu stören. Die neugebildeten Krebszellen häuften vermehrt Erbfehler an und bildeten vielfache Chromosomensätzen, was zu bizarrem Aussehen führte und Zellen mit riesigen Zellkernen entstehen ließ. Tumoren mit diesen „Monsterzellen“ schrumpften, selbst wenn die Krebszellen bereits vorher resistent gegenüber der Therapie geworden waren, so zeigte die Studie in Mäusen. Die abnormale Zellteilung und ungleiche Chromosomenverteilung sorgte bei den Zellen dafür, dass ihre Apoptose-Maschinerie wieder funktionierte. Dies war jedoch nur in Tumoren mit einem hohen PRC1-Spiegel der Fall.
„Frühere Ergebnisse ließen zunächst vermuten, dass die hier eingesetzte Wirkstoffkombination bei Patienten mit Ewing-Sarkom eher nicht vielversprechend ist“, sagt Grünewald. „Die neuen Daten geben uns jetzt Hoffnung, dass sie bei Patienten mit einer hoher PRC1-Aktivität jedoch durchaus wirksam sein könnten. Es zeigt einmal mehr, wie wichtig es ist, Krebstherapien an den molekularen Hintergrund des Tumors anzupassen.“ Ob sich der PRC1-Spiegel als zuverlässiger Biomarker eignet, wollen die Wissenschaftler jetzt mit weiteren Untersuchungen überprüfen und bei Erfolg in einer klinischen Studie weiterentwickeln.
Originalveröffentlichung
Weitere News aus dem Ressort Wissenschaft





















Meistgelesene News












Weitere News von unseren anderen Portalen






















Zuletzt betrachtete Inhalte
Dysmelie

Multifunktionales Pflaster zur Wundheilung - Film aus Biomolekülen haftet auf sensiblem Gewebe und setzt Wirkstoffe frei
