Maschinelles Lernen beschleunigt Materialsimulationen
Schnellere und genauere Methoden kommen verschiedenen Anwendungen von Energiespeichern bis zu Medikamenten zugute
Erforschung, Entwicklung und Herstellung neuer Materialien hängen entscheidend von schnellen und zugleich genauen Simulationsmethoden ab. Maschinelles Lernen, bei dem künstliche Intelligenz (KI) selbstständig neues Wissen erwirbt und anwendet, wird es künftig ermöglichen, komplexe Materialsysteme rein virtuell zu entwickeln. Wie das funktioniert und welche Anwendungen davon profitieren, erklärt ein Forscher des Karlsruher Instituts für Technologie (KIT) gemeinsam mit Kollegen aus Göttingen und Toronto in einem Artikel in der Zeitschrift Nature Materials.
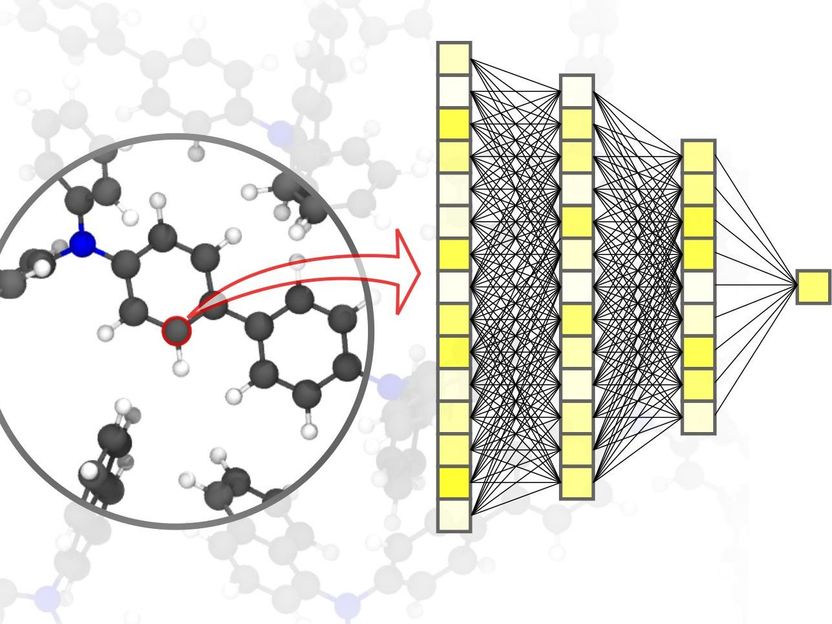
Neuronale Netze ermöglichen präzise Materialsimulationen – bis hinunter auf die Ebene einzelner Atome.
Pascal Friederich, KIT
Digitalisierung und Virtualisierung gewinnen in den verschiedensten wissenschaftlichen Disziplinen immer mehr an Bedeutung. Dies gilt auch für die Materialwissenschaft: Erforschung, Entwicklung und Herstellung neuer Materialien hängen entscheidend von schnellen und zugleich genauen Simulationsmethoden ab. Davon wiederum profitieren ganz unterschiedliche Anwendungen – von effizienten Energiespeichern, wie sie bei der Nutzung erneuerbarer Energien unverzichtbar sind, bis hin zu neuen Medikamenten, deren Entwicklung das Verständnis komplexer biologischer Vorgänge voraussetzt. Methoden der KI und des Maschinellen Lernens können Materialsimulationen entscheidend voranbringen. „Gegenüber herkömmlichen Simulationsmethoden, die auf klassischen oder quantenmechanischen Rechnungen basieren, lässt sich mit speziell auf Materialsimulationen zugeschnittenen neuronalen Netzen ein deutlicher Geschwindigkeitsvorteil erreichen“, erklärt der Physiker und KI-Experte Professor Pascal Friederich, Leiter der Forschungsgruppe AiMat – Artificial Intelligence for Materials Sciences am Institut für Theoretische Informatik (ITI) des KIT. „Schnellere Simulationssysteme werden es Wissenschaftlerinnern und Wissenschaftlern in den kommenden Jahren ermöglichen, größere und komplexere Materialsysteme rein virtuell zu entwickeln, sie bis auf die atomare Ebene hinunter zu verstehen und zu optimieren.“
Hohe Präzision vom Atom bis zum Werkstoff
In einem in der Zeitschrift Nature Materials veröffentlichten Artikel gibt Pascal Friederich, der auch als assoziierter Gruppenleiter im Bereich Nanomaterials by Information-Guided Design am Institut für Nanotechnologie (INT) des KIT tätig ist, gemeinsam mit Forschern der Universität Göttingen und der University of Toronto einen Überblick über die grundlegenden Prinzipien des für Materialsimulationen eingesetzten Maschinellen Lernens, den Datenerfassungsprozess sowie aktive Lernverfahren. Algorithmen für Maschinelles Lernen ermöglichen Künstlicher Intelligenz, die eingegebenen Daten nicht nur zu verarbeiten, sondern in großen Datensätzen Muster und Korrelationen zu finden, daraus zu lernen und selbstständig Vorhersagen und Entscheidungen zu treffen. Bei Materialsimulationen kommt es darauf an, eine hohe Präzision über verschiedene Zeit- und Größenskalen – vom Atom bis zum Werkstoff – zu erreichen und zugleich die Rechenkosten zu begrenzen. In ihrem Artikel gehen die Wissenschaftler auch auf verschiedene aktuelle Anwendungen ein, wie kleine organische Moleküle und große Biomoleküle, strukturell ungeordnete feste, flüssige und gasförmige Materialien sowie komplexe kristalline Systeme – beispielsweise metallorganische Gerüstverbindungen, die sich zur Gasspeicherung oder zur Stofftrennung, für Sensoren oder für Katalysatoren einsetzen lassen.
Noch mehr Tempo mit hybriden Methoden
Um die Möglichkeiten der Materialsimulationen zukünftig noch zu erweitern, schlagen die Forschenden aus Karlsruhe, Göttingen und Toronto vor, hybride Methoden zu entwickeln: Diese verbinden Verfahren des Maschinellen Lernens (ML) und der Molekularen Mechanik (MM) miteinander. MM-Simulationen bedienen sich sogenannter Kraftfelder, um die auf jedes einzelne Teilchen wirkenden Kräfte zu berechnen und damit Bewegungsabläufe vorherzusagen. Die Ähnlichkeit der ML- und MM-Potenziale erlaubt eine enge Integration mit variablen Übergangsbereichen. Solche hybriden Methoden könnten künftig beispielsweise die Simulation großer Biomoleküle oder enzymatischer Reaktion noch einmal deutlich beschleunigen.
Originalveröffentlichung

Weitere News aus dem Ressort Wissenschaft






















































