Pharmaforschung: Wenn sich Wirkstoff und Zielprotein „umarmen“
Bindungskinetik von Kinase-Hemmern untersucht
Wie sich die Passform bestimmter Wirkstoffe optimieren lässt, damit sie länger an ihre Zielproteine binden und damit eine stärkere pharmakologische Wirkung entfalten, haben jetzt Wissenschaftler der Goethe-Universität Frankfurt zusammen mit Kollegen aus Darmstadt, Heidelberg, Oxford und Dundee (UK) am Beispiel so genannter Kinase-Hemmstoffe untersucht. Solche Stoffe werden vielfach in der Krebstherapie eingesetzt. Das Ergebnis: Besonders lange dauert die „Umarmung“ von Hemmstoff und Protein, wenn sich das Protein an den Hemmstoff „anschmiegt“. Künftig wollen die Forscher mit Computersimulationen die Bindedauer von Substanzen vorhersagen.
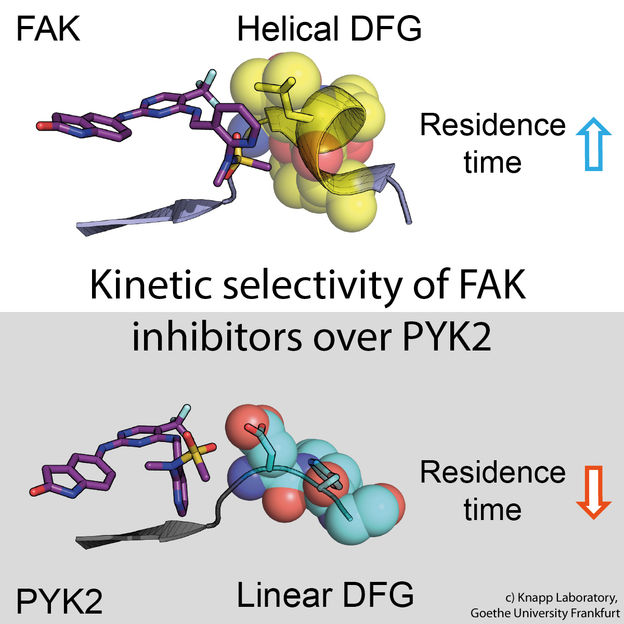
Oben: Lange Bindungsdauer. Ein Inhibitor (links: Stäbchenmodell) bindet an das Signalprotein FAK (rechts: ein Teil des FAK-Proteins dargestellt als Kalottenmodell mit Halbkugeln). Die strukturellen Veränderungen in FAK verursachen wasserabweisende Kontakte (gelb, so genanntes DFG-Motiv) und eine lang anhaltende Bindung. Unten: Kurze Bindungsdauer. Das PYK2-Signalprotein ändert seine Struktur nicht, was zu einer schnellen Dissoziation des Inhibitors führt.
Knapp Laboratory, Goethe-Universität Frankfurt
Viele Krebsmedikamente blockieren in Krebszellen Signale, mit deren Hilfe sich entartete Zellen unkontrolliert vermehren und aus dem Gewebeverband herauslösen. So führt zum Beispiel die Blockade des Signalproteins FAK, einer sogenannten Kinase, dazu, dass bestimmte Brustkrebszellen weniger beweglich werden und somit weniger stark metastasieren. Das Problem: Wenn FAK durch einen Hemmstoff blockiert wird, wird das nahe verwandte Signalprotein PYK2 viel aktiver und übernimmt so einen Teil der Aufgaben von FAK. Ideal wäre daher ein Hemmstoff, der in gleicher Weise sowohl FAK wie auch PYK2 möglichst langanhaltend inhibiert.
Ein internationales Team um den Pharmakochemiker Prof. Stefan Knapp von der Goethe-Universität hat eine Reihe eigens synthetisierter FAK-Hemmstoffe untersucht. Alle Hemmstoffe banden ungefähr gleich schnell an das FAK-Signalprotein. Sie unterschieden sich jedoch in der Dauer der Bindung: Der wirksamste Hemmstoff blieb am längsten mit dem FAK-Signalprotein verbunden.
In biochemischen und molekularbiologischen Analysen sowie Computersimulationen fand das Forschungsteam heraus, dass hierfür die Art der Wechselwirkung zwischen FAK-Signalprotein und Hemmstoff verantwortlich ist. Durch die Bindung des Wirkstoffs verändert das FAK-Signalprotein seine Form und bildet an einer der Kontaktstellen eine bestimmte, wasserabweisende Struktur aus. Diese veränderte (induzierte) FAK-Struktur bindet besonders gut an eine ebenfalls wasserabweisende Struktur des Hemmstoffs, vergleichbar einer innigen Umarmung.
Das Schwesterprotein PYK2 hingegen bleibt vergleichsweise steif, und obwohl der wirksamste FAK-Hemmstoff auch PYK2 blockierte, war sein Effekt hier deutlich schwächer.
Den Forscher:innen gelang es, das Bindungsverhalten der Inhibitoren in Computersimulationen zu modellieren und so eine Methode zu entwickeln, mit deren Hilfe sich künftig in der pharmazeutischen Forschung Wirkstoffkandidaten optimieren lassen.
Prof. Stefan Knapp erklärt: „Weil wir jetzt die molekularen Mechanismen der Interaktion von potenten Hemmstoffen dieser zwei Kinasen besser verstanden haben, hoffe wir, künftig anhand von Computersimulationen die Verweildauer potenzieller Wirkstoffe besser vorhersagen zu können. Die Verweildauer von Wirkstoffen wurde bisher nur wenig beachtet. Diese Eigenschaft hat sich jedoch als wichtiger Parameter für die Entwicklung von effektiven Wirkstoffen entpuppt, die nicht nur spezifisch, sondern auch langanhaltend ein oder mehrere Zielproteine – wie im Fall von FAK und PYK2 – hemmen sollen.“
Originalveröffentlichung
Benedict-Tilman Berger, Marta Amaral, Daria B. Kokh, Ariane Nunes-Alves, Djordje Musil, Timo Heinrich, Martin Schröder, Rebecca Neil, Jing Wang, Iva Navratilova, Joerg Bomke, Jonathan M. Elkins, Susanne Müller, Matthias Frech, Rebecca C. Wade, Stefan Knapp; "Structure-kinetic relationship reveals the mechanism of selectivity of FAK inhibitors over PYK2"; Cell Chemical Biology; 2021

Weitere News aus dem Ressort Wissenschaft




















Diese Produkte könnten Sie interessieren
Pharmaceutical Substances von Thieme Verlag
Entdecken Sie industrielle Synthesewege für 2.600 APIs
Ihr Recherchetool für Synthesen, Patente und Anwendungen – Pharmaceutical Substances

KNAUER IJM NanoScaler von KNAUER
Effiziente Formulierung von Lipid-Nanopartikeln für RNA-basierte Therapien
Optimieren Sie die Wirkstoffverkapselung von 1 ml bis zu Hunderten von Millilitern mit minimalem Wirkstoffeinsatz


































