3D-Landkarten der Genaktivität
Mithilfe eines dreidimensionalen Computermodells lässt sich künftig schnell ermitteln, welche Gene jeweils in welchen Zellen und an welcher Stelle eines Organs aktiv sind. Ein Team um Nikolaus Rajewsky, Berlin, und Nir Friedman, Jerusalem, stellt die neue Methode und darauf basierende Erkenntnisse in „Nature“ vor.

NovoSpaRc ermöglicht ein dreidimensionales Puzzle der Genexpression.
© Lior Friedman
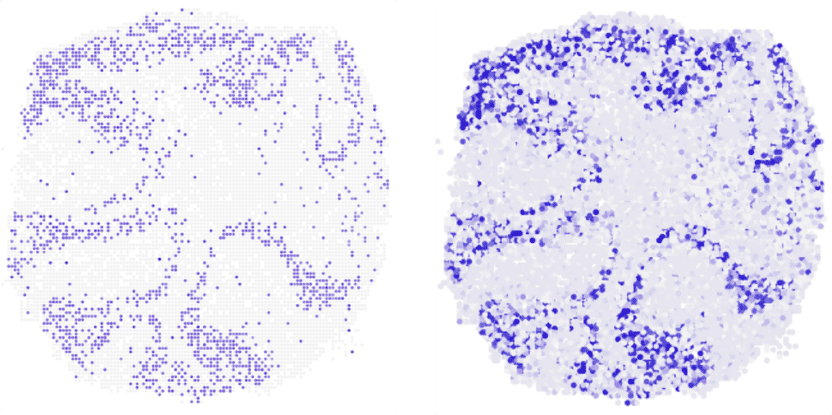
Wo im Mäuse-Kleinhirn wird gerade das Gen Neurod1 abgelesen? Der Längsschnitt zeigt, dass novoSpaRc (rechts) diese Frage anhand vorliegender Einzelzell-Sequenzierdaten sehr zuverlässig beantworten kann. Links zum Vergleich das Ergebnis aus Experimenten.
© AG N. Rajewsky, MDC

Professor Nikolaus Rajewsky ist ein Visionär: Er möchte verstehen, was in den menschlichen Zellen während einer Erkrankung geschieht – um schon die ersten Veränderungen in der Zelle zu erkennen und gezielt zu behandeln. „Dazu müssen wir nicht nur die Aktivität des Genoms in einzelnen Zellen entschlüsseln, sondern sie vor allem auch räumlich innerhalb eines Organs verfolgen“, sagt der Wissenschaftliche Direktor des Berliner Instituts für Medizinische Systembiologie (BIMSB) am Max-Delbrück-Centrum für Molekulare Medizin (MDC) in Berlin. Zum Beispiel sei die räumliche Anordnung von Immunzellen bei einer Krebserkrankung (die „Mikroumgebung“) extrem wichtig, um genau zu diagnostizieren und die beste Therapie zu wählen. Ganz generell fehle ein systematischer Ansatz, die (Patho-)Physiologie eines Gewebes molekular zu erfassen und zu verstehen.
Karten für ganz verschiedene Gewebetypen
Mit einer großen neuen Studie, die jetzt im Fachblatt „Nature“ erscheint, ist Rajewsky seinem Ziel einen großen Schritt nähergekommen. Gemeinsam mit Professor Nir Friedman von der Hebrew University in Jerusalem, Dr. Mor Nitzan von der Harvard University im US-amerikanischen Cambridge und Dr. Nikos Karaiskos, einem Projektleiter aus seiner eigenen Arbeitsgruppe „Systembiologie von Gen-regulatorischen Elementen“, ist es dem Wissenschaftler gelungen, mithilfe eines speziellen Algorithmus eine Art räumliche Landkarte der Genexpression für einzelne Zellen in ganz verschiedenen Gewebetypen zu erstellen: in der Leber und dem Darmepithel von Säugetieren, in Embryonen von Taufliegen und Zebrafischen sowie in Teilen des Kleinhirns und der Niere. „Manchmal reicht rein theoretische Wissenschaft, um in einem hochrangigen Wissenschaftsjournal zu publizieren - ich glaube, das wird in der Zukunft noch häufiger vorkommen. Wir müssen noch viel mehr in Machine Learning und Künstliche Intelligenz investieren“, sagt Nikolaus Rajewsky.

„Anhand der im Computer erstellten Karten können wir nun genau verfolgen, ob ein bestimmtes Gen in den Zellen eines Gewebeteils aktiv ist oder nicht“, erklärt Karaiskos, theoretischer Physiker und Bioinformatiker, der den Algorithmus gemeinsam mit Dr. Mor Nitzan von der Universität Harvard maßgeblich entwickelt hat. „Das war ohne unser Modell, das wir novoSpaRc genannt haben, in dieser Form nicht möglich.“
Räumliche Informationen gingen bisher verloren
Erst seit wenigen Jahren können Forscher überhaupt in großem Maßstab und mit hoher Präzision nachvollziehen, welche Informationen einzelne Zellen in einem Organ oder Gewebe gerade aus dem Erbgut abrufen. Ermöglicht haben das neue Sequenziermethoden, zum Beispiel die Multiplex-RNA-Sequenzierung, mit deren Hilfe sich eine Vielzahl von RNA-Molekülen gleichzeitig untersuchen lässt. RNA wird in der Zelle gebildet, wenn Gene aktiv werden und anhand ihres Bauplans Proteine entstehen. Rajewsky erkannte das Potenzial der Einzelzell-Sequenzierung früh und etablierte sie in seinem Labor.
„Eine Voraussetzung für die Technologie ist allerdings, dass das untersuchte Gewebe in einzelne Zellen zerlegt werden muss“, sagt Rajewsky. Dadurch gehen jedoch wertvolle Informationen verloren: Niemand weiß mehr, wo im Gewebe die Zelle saß, deren Genaktivität man ermittelt hat. Rajewsky und Friedmann suchten daher nach einer Möglichkeit, um anhand von Daten aus Einzelzell-Sequenzierungen ein mathematisches Modell zu entwickeln, mit dessen Hilfe sich das räumliche Muster der Genexpression für das gesamte Erbgut auch in komplex aufgebauten Geweben errechnen lässt.
Ein erster Schritt war den Teams um Rajewsky und Dr. Robert Zinzen, der ebenfalls am BIMSB arbeitet, bereits vor zwei Jahren gelungen. Im Fachblatt „Science“ hatten sie ein virtuelles Modell eines Taufliegen-Embryos vorgestellt. Es zeigte in bis dahin ungekannter räumlicher Auflösung, in welchen Zellen welche Gene jeweils aktiv sind. Gelungen war die Genkartierung mit der Hilfe von 84 Markergenen, von denen man durch in-situ-Experimente wusste, wo in dem eiförmigen Embryo die Gene zu einem bestimmten Zeitpunkt jeweils aktiv sind. Dass ihr Modell funktionierte, bestätigten die Forscherinnen und Forscher mit weiteren aufwändigen in-situ-Experimenten an lebenden Taufliegen-Embryonen.
Eine Art Puzzle mit Zigtausenden Teilen und Farben
„Bei dem Modell haben wir allerdings für jede Zelle einzeln rekonstruiert, wo sie verortet war“, sagt Karaiskos. Er war sowohl bei der „Science“-Studie als auch bei der aktuellen „Nature“-Studie einer der Erstautoren. „Das war damals möglich, weil wir es mit einer deutlich geringeren Zahl von Zellen und Genen zu tun hatten. Dieses Mal wollten wir dagegen wissen, ob wir komplexe Gewebe rekonstruieren können, wenn uns vorab kaum Informationen vorliegen. Können wir die Prinzipien nachvollziehen, nach denen die Genaktivität in komplexen Geweben organisiert und gesteuert ist?“ Die Grundannahme für den Algorithmus war: Wenn Zellen benachbart sind, ähnelt sich ihre Genaktivität. Sie lesen also ähnlichere Informationen aus ihrem Erbgut ab, als Zellen, die weiter voneinander entfernt sind.
Um diese Hypothese zu prüfen, haben die Forscherinnen und Forscher auf vorhandene Daten zurückgegriffen. Für die Leber, Niere und das Darmepithel gab es keine zusätzlichen Informationen. Einige wenige Markergene hatte die Gruppe zuvor mithilfe rekonstruierter Gewebeproben gewonnen. In einem Fall waren es gerade einmal zwei Markergene gewesen.
„Es war wie ein sehr großes Puzzle mit einer riesigen Anzahl – vielleicht 10.000 – verschiedener Farben“, versucht Karaiskos die kniffelige Aufgabe zu beschreiben, vor die er bei der Berechnung des Modells gestellt war. „Wenn das Puzzle richtig gelöst ist, ergeben all diese Farben jeweils für sich genommen eine ganz bestimmte Form oder ein spezielles Muster.“ Jedes Puzzleteil steht dabei für eine einzelne Zelle des untersuchten Gewebes und jede Farbe für ein aktives Gen, das gerade mit einem RNA-Molekül abgelesen wird.
Die Methode funktioniert unabhängig vom Sequenzierverfahren
„Wir verfügen somit jetzt über eine Methode, mit der man anhand der Daten einer Einzelzell-Sequenzierung im Computer ein virtuelles Modell des untersuchten Gewebes erstellen kann – und zwar unabhängig davon, welches Sequenzierverfahren genutzt wurde“, sagt Karaiskos. „Bereits vorhandene Informationen über die räumliche Lage einzelner Zellen lassen sich in das Modell einspeisen und können es so weiter verfeinern.“ Mithilfe von novoSpaRc lasse sich dann für jedes bekannte Gen erfragen, wo genau im Gewebe die Erbanlage aktiv sei und in ein Protein übersetzt werde.
Derzeit versuchen Karaiskos und seine Kollegen am Berliner Institut für Medizinische Systembiologie zudem, das Modell so zu nutzen, dass sie damit bestimmte Entwicklungsvorgänge in Geweben oder ganzen Organismen zurückverfolgen und auch vorhersagen können. Allerdings, so räumt der Wissenschaftler ein, bleibe noch abzuwarten, ob es womöglich bestimmte Gewebe gebe, auf die sich der Algorithmus von novoSpaRc nicht anwenden lasse. Womöglich würde er sich darüber sogar freuen: Es käme ein neues herausforderndes Puzzle auf ihn zu.
Originalveröffentlichung

Weitere News aus dem Ressort Wissenschaft























































