Software sagt voraus, an welchen Stellen Genschalter an die DNA binden
Bioinformatiker bieten preisgekröntes Programm als Open Source an
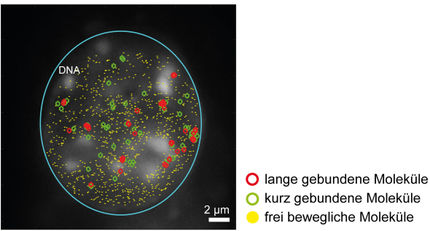
Nicht jedes der unzähligen Gene im Erbgut eines Lebewesens wird auch abgelesen. Sie können ein- und ausgeschaltet werden, zum Beispiel, indem bestimmte Proteine, sogenannte Transkriptionsfaktoren, an spezifischen Stellen im Genom andocken. Solche Transkriptionsfaktoren können bei der Entstehung und Behandlung von Krankheiten eine Rolle spielen. So sind beispielsweise einige psychische Erkrankungen oder die Entstehung mancher Tumore in der Lunge mit Mutationen von Bindungsstellen für Transkriptionsfaktoren verbunden.
„Ein experimenteller Nachweis solcher Bindungsstellen ist extrem aufwändig und teuer“, erklärt der Bioinformatiker Dr. Jens Keilwagen vom Julius Kühn-Institut. Mit Prof. Dr. Stefan Posch und Dr. Jan Grau von der MLU hat er deswegen eine Software entwickelt, die Bindungsstellen für Transkriptionsfaktoren in verschiedenen Gewebearten vorhersagen kann. Mit einem Prototyp dieses Programms hat das Bioinformatiker-Team im März 2017 die weltweite „ENCODE-DREAM in vivo Transcription Factor Binding Site Prediction Challenge“ gewonnen.
Nun haben die Forscher ihr Programm weiter optimiert und das Ergebnis ihrer Arbeit im Open-Access-Journal „Genome Biology“ veröffentlicht. Damit ermöglichen die Informatiker anderen Wissenschaftlern freien Zugang zu der weiterentwickelten Open-Source-Software. „In der Challenge reichte die Zeit nicht, um das Programm in der Tiefe zu evaluieren“, sagt Keilwagen. Das haben die Informatiker jetzt nachgeholt. So konnten die Wissenschaftler überflüssige Messungen entfernen und außerdem die wichtigsten Faktoren für die Vorhersage von Bindungsstellen identifizieren. „Entscheidend sind Zugänglichkeit zum DNA-Strang und Erkennungsmotiv“, erklärt Dr. Jan Grau von der MLU. „Die DNA ist in unterschiedlichen Gewebetypen unterschiedlich verpackt, was bestimmt, wie zugänglich die DNA für Transkriptionsfaktoren ist.“ Ein Erkennungsmotiv beschreibt Abfolgen von Nukleinbasen, die von einem bestimmten Transkriptionsfaktor besonders häufig gebunden werden.
Das nun veröffentlichte Programm ist schneller und anwenderfreundlicher als der Prototyp und hat so Potenzial als Werkzeug in der Grundlagenforschung. Bei einigen Transkriptionsfaktoren ermöglicht es bereits eine verlässliche Vorhersage von Bindungsstellen. Durch weitere Verbesserungen könnten langfristig, so die Hoffnung der Informatiker, aufwendige experimentelle Nachweise durch eine bioinformatische Vorhersage ersetzt werden.
Bisher ist das Programm auf Vorhersagen für menschliche Zellen beschränkt. Die Bioinformatiker von JKI und MLU planen aber bereits die Anwendung auf Pflanzenzellen. „Damit könnte man zum Beispiel prüfen, wie eine Pflanze auf Stress reagiert – wo binden dann welche Transkriptionsfaktoren, und was lösen sie dabei aus?“, so Keilwagen.
Originalveröffentlichung
Weitere News aus dem Ressort Wissenschaft




















Diese Produkte könnten Sie interessieren

Limsophy LIMS von AAC Infotray
Optimieren Sie Ihre Laborprozesse mit Limsophy LIMS
Nahtlose Integration und Prozessoptimierung in der Labordatenverwaltung
GUS-OS Suite von GUS
Ganzheitliche ERP-Lösung für Unternehmen der Prozessindustrie
Integrieren Sie alle Abteilungen für nahtlose Zusammenarbeit


































