Seltene Erkrankung enthüllt unbekannten Mechanismus der Fettverdauung
Patienten mit CDDs (congenital diarrheal disorders), einer Gruppe seltener, weitgehend unerforschter Erbkrankheiten, leiden häufig bereits bei der Geburt an schweren, oft lebensbedrohlichem Durchfall. Wissenschaftler des LBI-RUD und des CeMM, zusammen mit der Medizinischen Universität Innsbruck und dem University Medical Center Utrecht haben nun die molekularen Mechanismen der Erkrankungen erforscht. Dabei sind sie auf ein Protein gestoßen, das eine entscheidende Rolle in der Fettverdauung spielt.
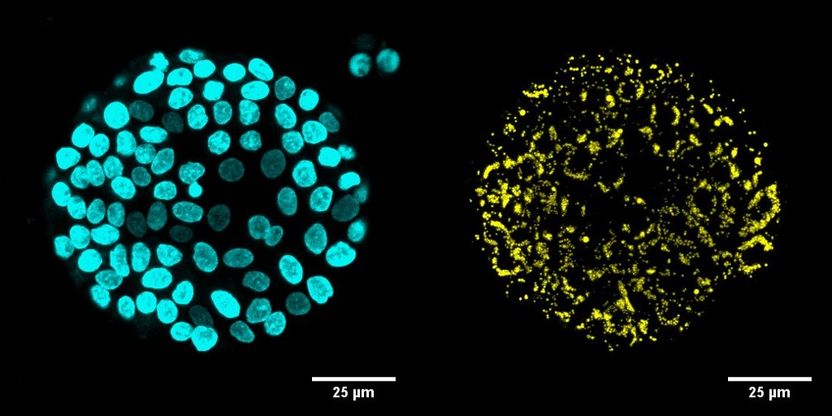
Immunofluoreszente Mikroskopieaufnahmen von Organoiden mit DAPI-(blau) und LD540-(gelb) Färbung von einem DGAT1-defizienten Patienten
Jorik M van Rijn
Damit Fett aus unserer Nahrung aufgenommen werden kann, muss es eine Reihe komplexer biochemischer Reaktionsketten durchlaufen. Die Fettmoleküle, bestehend aus einem Glycerin und drei Fettsäuren (daher auch Triglyceride genannt), müssen zunächst in ihre Bestandteile zerlegt werden um von den Zellen der Darmschleimhaut aufgenommen zu werden. In diesen Zellen werden die Triglyceride wiederhergestellt und in kleine Transportpartikel verpackt, die in den Blutkreislauf entlassen werden. Kommt es in diesen Prozessen der Fettverdauung zu Störungen, kann das verheerende Auswirkungen haben.
Dies hat sich bei zehn Kindern aus sechs Familien gezeigt, die seit der Geburt an CDD und damit an schwerem Durchfall und/oder Erbrechen litten. Nachdem sich eine Reihe konventioneller Therapien als wirkungslos erwies, wurde schließlich Kaan Boztug, Direktor des Ludwig Boltzmann Institute for Rare and Undiagnosed Diseases (LBI-RUD) sowie zugehöriger Forschungsgruppenleiter des CeMM Forschungszentrum für Molekulare Medizin der Österreichischen Akademie der Wissenschaften, hinzugezogen. In Kollaboration mit der Medizinischen Universität Innsbruck, dem University Medical Center Utrecht und der Ankara University School of Medicine führten die Wissenschaftler DNA-Sequenzierungen mit dem Erbgut der Patienten durch, und stießen auf Mutationen in dem Gen für das Protein diacylglycerol-acyltransferase 1 (DGAT1).
DGAT1 ist ein Enzym, das für den letzten Schritt der Triglycerid-Bildung in den Zellen der Darmschleimhaut verantwortlich ist. Die Forscher konnten nachweisen, dass die bei den Patienten gefundenen Mutationen des DGAT1 Gens zu einer reduzierten oder vollständig eingestellten Proteinproduktion in den Zellen führte. In weiteren Experimenten zeigte sich, dass diese Zellen nicht in der Lage waren, Fette für ihren Stoffwechsel richtig zu nutzen. Darüber hinaus gelang es den Forschern, Darm-Organoide – miniaturisierte und vereinfachte Strukturen mit Organ-ähnlichen Eigenschaften – aus Patientenbiopsien zu züchten und die Auswirkungen des Gendefekts auch hier zu beobachten.
„Darm-Organoide sind ausgezeichnete Modellsysteme, um gastrointestinale Erkrankungen zu erforschen“, erklärt Rico Ardy, PhD-Student am LBI-RUD und CeMM und Co-Erstautor der Studie. „Gemeinsam mit den anderen experimentellen Ansätzen haben sie uns ermöglicht, die genetische Ursache und die molekularen Mechanismen dieser seltenen Erkrankung aufzuklären. Unsere Ergebnisse zeigen ganz allgemein, welche Rolle DGAT1 in einem gesunden Fettstoffwechsel spielt.“
Die Studie ist ein weiterer Beweis dafür, dass genetische Analysen für Patienten mit angeborenen Erkrankungen einen wichtigen Beitrag für die Entwicklung einer passenden Pflege und Therapie darstellen. Die Arbeit zeigt darüber hinaus die generelle Bedeutung der Forschung an seltenen Erkrankungen, die in vielen Fällen nicht nur den betroffenen Patienten hilft, sondern auch neue Erkenntnisse über die menschliche Biologie liefert.
„Seltene Erkrankungen zu erforschen enthüllen oft fundamentale Prozesse der menschlichen Physiologie und helfen uns dabei, Therapien zu finden in Fällen, wo die konventionellen Ansätze versagen“ fügt Kaan Boztug hinzu. „Die Patienten mit DGAT1-Defizienz wurden zunächst auf eine fettfreie Diät gesetzt, mit der sich die Verdauung der Patienten normalisierte. Die Forscher des LBI-RUD arbeiten unterdessen an spezifischeren, personalisierten Therapien um solche Erbkrankheiten in der Zukunft gezielter zu behandeln.“
Originalveröffentlichung

Weitere News aus dem Ressort Wissenschaft





















Meistgelesene News











Weitere News von unseren anderen Portalen





















Zuletzt betrachtete Inhalte
Verband_(Medizin)
Mykoplasmen
Nano-Sicherheit wird verstärkt erforscht
Sanofi will Johnson & Johnson bei Impfstoff-Produktion helfen
