Künstlicher Gendefekt enthüllt möglichen Angriffspunkt für Erbkrankheit
Fanconi-Anämie, eine seltene Erbkrankheit, wird durch defekte DNA-Reparaturgene in den Zellen der Patienten verursacht und führt zu Knochenmarksversagen, Entwicklungsstörungen und erhöhtem Krebsrisiko. Mit Hilfe genomweiter genetischer Methoden erzeugten WissenschaftlerInnen am CeMM systematisch künstliche Gendefekte in FA-Zellen und fanden so ein Gen, das –wenn ausgeschaltet –den Effekt der Krankheit in den Zellen aufhebt. Das dazugehörige Protein stellte sich als vielversprechendes Ziel für eine potentielle Therapie der Erbkrankheit heraus.

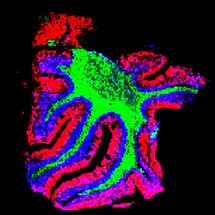
Karyotyp von HAP1 Zellen ohne FANCC-Gen, für 24 h mit MMC behandelt. QR = quadri radial Chromosomenvernetzungen
CeMM/KaryoLogic
Die Fähigkeit, DNA zu reparieren, ist für einen gesunden Organismus unverzichtbar. Zehntausende Schäden ereignensich jeden Tag im Erbgut der Zellen, es verwundert daher nicht, dass die Evolution eine ganze Palette an Reparaturmechanismen hervorgebracht hat, die es den Zellen ermöglicht,die Schäden möglichst schnell zu beheben. Die Bedeutung dieser Prozesse wird offensichtlich, wenn sie versagen: Die Zellen von Patienten mit Fanconi-Anämie (FA) sind nicht in der Lage, Vernetzungen der DNA -eine bestimmte Art von DNA-Schäden –zu reparieren. Die Folgen sind verheerend, neben zahlreichen anderen Symptomen entwickeln sich in den meisten FA-Patienten Tumore. Bisher gibt es keine kurative Behandlung der Krankheit.
DNA-Schäden und die komplexen Reparaturmechanismen sind der Forschungsschwerpunkt von Joanna Loizou, Forschungsgruppenleiterin am CeMM Forschungszentrum für Molekulare Medizin der Österreichischen Akademie der Wissenschaften, und die Suche nach neuen Therapieansätzen für die Fanconie-Anämie ist eines ihrer Ziele. In ihrer neuesten Studie haben WissenschaftlerInnen aus ihrem Team nach zusätzlichen Genen gesucht, die mit den defekten DNA-Reparaturgenen der FA-Patienten interagieren,und die für die Ausprägung der Krankheit essentiell sind. Denn, so die Hypothese, wenn man diese Gene ebenfalls zerstört, ließe sich die Fähigkeit der Zelle, DNA-Vernetzungen zu reparieren, wiederherstellen.Die Forschungsarbeit erfolgte in Kooperation mit WissenschaftlerInnen der University of Cambridge, dem Leiden University Medical Center, der University of California, der University of Toronto und der Gruppe von Jörg Menche am CeMM.Was wie ein Widerspruch klingt, ist ein erprobtes Konzept: Durch zusätzliches Ausschalten weiterer Gene bei einem bereits vorhandenem Gendefekt kann es zu einer Umlagerung der hochkomplexen molekularen Netzwerke der Zelle kommen. Mechanismen wie die DNA-Reparatur können auf diesem Weg wieder funktionsfähig gemacht werden. Einen solcher Ansatz wurde auch in dieser Studie von den WissenschaftlerInnen -mit Georgia Velimezi, ehemalige Postdoktorantin und Lydia Garcia-Robinson, PhD Studentin in Loizou ́s Gruppe als Co-Erstautorinnen -angewandt. Mit einer genomweiten Analyse, in der jedes Gen von FA-Zellen einzeln ausgeschaltet wurde, fanden sie tatsächlich eines, das die Fähigkeit zur Reparatur von DNA-Vernetzungen wiederherstellte. Das gefundene Gen codiert für ein Enzym, USP48, das einen wichtigen Regulationsfaktor für Proteine entfernen kann. Wenn USP48 in FA-Zellen nicht mehr vorhanden ist, reagieren die Zellen viel weniger empfindlich auf DNA-schädigende Substanzen und können die Schäden viel besser reparieren. „Unsere Ergebnisse zeigen, dass die Inaktivierung von USP48 die chromosomale Stabilität der FA-Zellen erhöht“ erklärt Joanna Loizou. „Das unterstreicht die Bedeutung dieses Enzyms in der Kontrolle der DNA-Reparatur und macht es zu einem vielversprechenden Ziel für Wirkstoffe. Wenn es gelänge, gezielt inhibierende Moleküle gegen USP48 zu entwickeln, wäre das ein völlig neuer potentieller Ansatz um die verheerenden Symptome von FA-Patienten zu mildern.
Originalveröffentlichung
Georgia Velimezi, Lydia Robinson-Garcia, Francisco Muñoz-Martínez, Wouter W. Wiegant, Joana Ferreira da Silva, Michel Owusu, Martin Moder, Marc Wiedner, Sara Brin Rosenthal, Kathleen M. Fisch, Jason Moffat, Jörg Menche, Haico van Attikum, Stephen P. Jackson & Joanna I. Loizou; "Map of synthetic rescue interactions for the Fanconi anemia DNA repair pathway identifies USP48"; Nature Comm.; 2018
Weitere News aus dem Ressort Wissenschaft