Ein Mechanismus, der die Huntington-Krankheit abschwächt
Langfristig Hoffnung auf neue Therapie
Der Neurowissenschaftler Dr. David Vilchez und sein Team bei CECAD, dem Exzellenzcluster für Alternsforschung der Universität zu Köln, haben neue Erkenntnisse zu einem System gewonnen, das die neurodegenerative Huntington-Krankheit abschwächen könnte. Durch den jetzt entdeckten Mechanismus wird die Anhäufung von toxischen Proteinen verhindert, die für den Abbau von Hirnzellen bei der Krankheit verantwortlich sind.

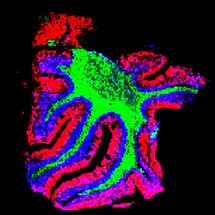
Der Modellorganismus C. elegans mit mutiertem Huntington Gen unter dem Mikroskop. Das Ausschalten des Schutz-Proteins UBR5 führte zu einem drastischen Anstieg des toxischen „Huntingtin“-Proteins in den Neuronen. Diese Anhäufungen sind auf dem Bild in grün zu erkennen.
Universität zu Köln
Die Huntington-Krankheit ist eine neurodegenerative Erkrankung, die zum Tod von Gehirnzellen und damit zu unkontrollierten Körperbewegungen, Sprachverlust und Psychosen führt. Mutationen des „Huntingtin“-Gens sind für die Krankheit verantwortlich, da sie dazu führen, dass sich das gleichnamige Protein „Huntingtin“ in giftiger Menge anhäuft. Diese Anhäufung führt zu Neurodegeneration und in den allermeisten Fällen zum Tod des Patienten spätestens zwanzig Jahre nach Ausbruch der Krankheit.
Um die Mechanismen der Huntington-Krankheit besser zu verstehen, untersuchten Vilchez und sein Team sogenannte induzierte pluripotente Stammzellen (iPS-Zellen) von Patienten mit der Huntington-Krankheit. Stammzellen sind in der Lage, sich in jeden Zelltyp – beispielsweise in Neuronen – zu differenzieren.

Die Stammzellen der Huntington-Patienten konnten erstaunlicherweise die sonst krankheitstypische Anhäufung des toxischen Proteins „Huntingtin“ vermeiden. Verantwortlich für diesen Erfolg zeigte sich ein Protein namens UBR5, das als Schutzmechanismus für die pluripotenten Stammzellen fungiert. UBR5 sorgt dafür, dass sich das schädliche „Huntingtin“-Protein wieder abbaut.
Das Kölner Team verglich unsterbliche iPS-Zellen von Huntington-Patientinnen und -Patienten und die daraus differenzierten Neuronen auf Unterschiede in ihrer Fähigkeit, eine Anhäufung des mutierten „Huntingtin“ zu vermeiden. Dabei stellte sich heraus, dass „Huntingtin“ durch das zelluläre Entsorgungssystem „Proteasom“ abgebaut werden kann. Doch gerade in den Neuronen der Patientinnen und Patienten war das so wichtige Entsorgungssystem defekt und „Huntingtin“ konnte nicht abgebaut werden.
Vilchez und sein Team fanden heraus, dass das Schutz-Protein UBR5 jedoch in den Stammzellen erhöht aufzufinden war. Das beschleunigte den Abbau von „Huntingtin“ in den Zellen. Sie kontrollierten auch, ob eine Herunterregulierung des UBR5-Spiegels, also weniger Schutz, zur giftigen Anhäufung von „Huntingtin“ in den Stammzellen führte. Tatsächlich stellten sie fest, dass das Senken von UBR5 zu einem massiven Anstieg in Anhäufungen von „Huntingtin“ in den Stammzellen führte. „Das war verblüffend“, sagt Vilchez. „Aus dem Nichts häuften die Zellen auf einmal große Mengen des toxischen Proteins an.“
Die Forscherinnen und Forscher überprüften ihre Ergebnisse auch an einem Modellorganismus, um die früheren Befunde noch einmal zu bestätigen. Beim Fadenwurm C. elegans testeten sie, ob sich durch eine Herunterregulierung des UBR5-Spiegels die Anhäufung von „Huntingtin“ in den Neuronen erhöht. Wieder führte das Senken von UBR5 zu einem massiven Anstieg der Anhäufung und neurotoxischen Wirkung des schädlichen Proteins in den Neuronen. Die Erhöhung von UBR5 hingegen blockierte die Anhäufung von „Huntingtin“ in den Modellorganismen.
Das Team testete auch, ob die Ergebnisse auf andere neurodegenerative Krankheiten zutreffen. „Wir haben den Mechanismus bei der amyotrophen Lateralsklerose (ALS) unter die Lupe genommen“, sagt Seda Koyuncu, eine Doktorandin am Labor von Vilchez und Hauptautorin der Publikation. „Aber unser Ergebnis ist sehr spezifisch für die Huntington-Krankheit“, erläutert Dr. Isabel Saez, eine weitere Hauptautorin und CECAD-Mitarbeiterin.
Auch wenn die Ergebnisse für die Entwicklung neuer Behandlungen und Medikamente wichtig sein könnten, gebe es noch keine Heilung für die Huntington-Krankheit. „Es ist nicht so, dass man etwas Neues entdeckt und dann gibt es sofort eine Heilung; es ist schwieriger. Aber in einigen Jahren könnte es eine Therapie geben“, sagt Saez. Bis dahin muss noch weiter geforscht werden. „Diese Ergebnisse tragen zu einem besseren Verständnis der Huntington-Krankheit bei und könnten langfristig ein Meilenstein in der Behandlung von Patienten sein“, sagt Vilchez.
Originalveröffentlichung
Weitere News aus dem Ressort Wissenschaft



















Diese Produkte könnten Sie interessieren

Antibody Stabilizer von CANDOR Bioscience
Protein- und Antikörperstabilisierung leicht gemacht
Langzeitlagerung ohne Einfrieren – Einfache Anwendung, zuverlässiger Schutz

DynaPro NanoStar II von Wyatt Technology
NanoStar II: DLS und SLS mit Touch-Bedienung
Größe, Partikelkonzentration und mehr für Proteine, Viren und andere Biomoleküle