Neue Software verarbeitet riesige Mengen an Einzelzelldaten
Umfängliche Analyse großer Genexpressionsdatensätze
Wissenschaftler des Helmholtz Zentrums München haben ein neues Programm entwickelt, das große Datensätze beherrschbar machen soll. Die Software mit dem Namen Scanpy ist beispielsweise ein Kandidat für die Auswertung des Human Cell Atlas Projekts.
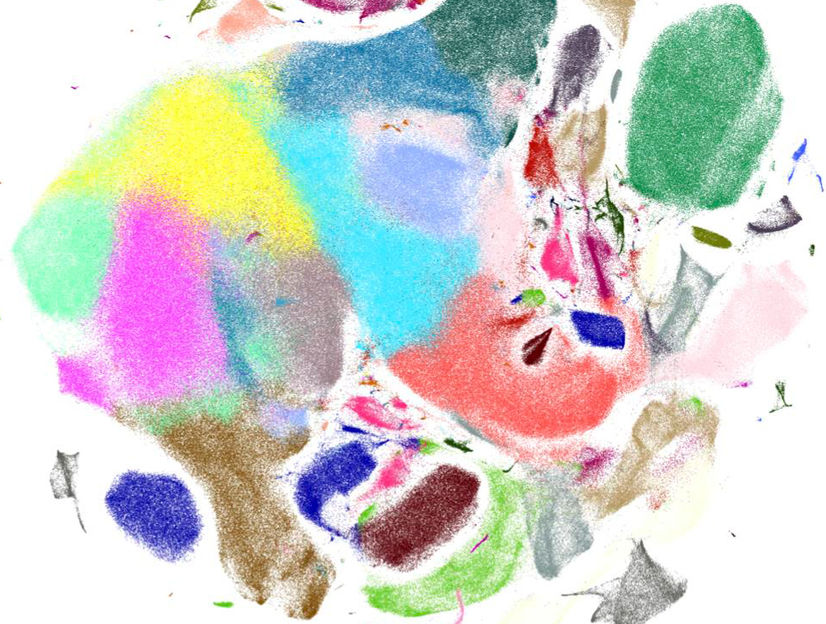
Visualisierung von Genexpressionsmustern muriner Gehirnzellen mit Scanpy.
Helmholtz Zentrum München
„Es geht um die Analyse von Genexpressionsdaten zahlreicher einzelner Zellen“, erklärt Erstautor Alex Wolf vom Institute of Computational Biology (ICB) des Helmholtz Zentrums München. Er hat Scanpy entwickelt, gemeinsam mit seinem Kollegen Philipp Angerer in der Machine Learning Gruppe von Institutsdirektor Prof. Dr. Dr. Fabian Theis, der neben seiner Position am Helmholtz Zentrum auch Professor für Mathematische Modelle biologischer Systeme an der TU München ist. „Die neue technische Möglichkeiten generieren um Größenordnungen mehr Daten mit dementsprechend höherer Information“, schildert Theis. „Allerdings war die historisch gewachsene Software-Infrastruktur zur Genexpressionsanalyse nicht auf die neuen Herausforderungen ausgelegt.“ Entsprechend groß sei hier der Bedarf nach neuen Analysemethoden.
Im Rennen für den Human Cell Atlas
Auch ein großes internationales Forschungsvorhaben könnte Theis zufolge von der Software profitieren. Unter dem Namen ‚Human Cell Atlas‘ tragen zahlreiche internationale Wissenschaftler eine Referenzdatenbank zusammen, in der die Genaktivität aller menschlichen Zelltypen erfasst ist. „Für dieses Projekt oder auch bei der immer häufiger werdenden Zusammenlegung von bestehenden Datensätzen ist es wichtig, eine skalierbare Software zu haben“, so Theis. Entsprechend sei Scanpy aktuell in der Auswahl für die Analysesoftware des Human Cell Atlas.
„Mit Scanpy publizieren wir die erste Software, die eine umfängliche Analyse großer Genexpressionsdatensätze mit einem breiten Spektrum aus Methoden des maschinellen Lernens und Statistik erlaubt“, beschreibt Alex Wolf den Fortschritt. „Bereits jetzt wird die Software in diversen Gruppen weltweit eingesetzt, insbesondere auch am Broad Institute von Harvard und dem Massachusetts Institute of Technology.“
Technologisch beschreitet die Anwendung neue Wege: Während entsprechende Biostatistik-Software traditionell in der Programmiersprache R geschrieben wurde, basiert Scanpy auf der Sprache Python, die die Machine Learning Community dominiert. Neu ist zudem, dass Graph-basierte Algorithmen das Herz von Scanpy bilden. Anstatt Zellen wie bisher üblich als Punkte im Koordinatensystem des Genexpressionsraums zu betrachten, verwenden die Algorithmen ein graphartiges Koordinatensystem. Das heißt, anstatt eine Zelle mit dem Expressionswert einiger Tausend Gene zu charakterisieren, wird sie einfach durch die Angabe ihrer nächsten Nachbarn charakterisiert – vergleichbar mit Verbindungen in sozialen Netzwerken. Wenn es zum Beispiel um die Identifikation von Zelltypen geht, verwendet Scanpy also die gleichen Algorithmen wie Facebook zur Identifikation von Communities.
Originalveröffentlichung
Weitere News aus dem Ressort Wissenschaft




















Diese Produkte könnten Sie interessieren

Limsophy LIMS von AAC Infotray
Optimieren Sie Ihre Laborprozesse mit Limsophy LIMS
Nahtlose Integration und Prozessoptimierung in der Labordatenverwaltung
GUS-OS Suite von GUS
Ganzheitliche ERP-Lösung für Unternehmen der Prozessindustrie
Integrieren Sie alle Abteilungen für nahtlose Zusammenarbeit

Meistgelesene News








Weitere News von unseren anderen Portalen






















Zuletzt betrachtete Inhalte

Luftige Cellulose aus dem 3D-Drucker - Wundermaterial mit großem Potenzial für die Medizin

Wie Zellen die Kurve kriegen - Solch universelle Prinzipien ermöglichen besseres Verständnis der Migration von Immun- und Krebszellen und ebnen den Weg für neue Behandlungsmöglichkeiten

Warum Tiefschlaf unser Gedächtnis fördert - Langsame Gehirnwellen machen Hirnrinde besonders aufnahmebereit

Analytica 2022 begeisterte die Laborbranche - analytica extended: digitale Verlängerung bis zum 8. Juli
Tecan präsentiert Freedom EVO auf der Biotechnica
