Method to profile and predict the behavior of proteins
An enzyme is a tiny, well-oiled machine. A class of proteins that are made up of multiple, interlocking molecular components, enzymes perform a variety of tasks inside each cell. However, precisely how these components work together to complete these tasks has long eluded scientists. But now, a team of researchers has found a way to map an enzyme's underlying molecular machinery, revealing patterns that could allow them to predict how an enzyme behaves—and what happens when this process disrupted.
In the latest issue of the journal Cell, a team of scientists led by Gladstone Institutes and University of California, San Francisco (UCSF) Investigator Nevan Krogan, PhD, Texas A&M University's Craig Kaplan, PhD, and UCSF Professor Christine Guthrie, PhD, describe a new technique—called the point mutant E-MAP (pE-MAP) approach—that gives researchers the ability to pinpoint and map thousands of interactions between each of an enzyme's many moving parts.
The researchers focused on a well-known enzyme—called RNA polymerase II (RNAPII)—and used the single-cellular yeast species S. cerevisiae as a model. Researchers had previously mapped the physical structure of RNAPII, but not how various parts of the enzyme work with other proteins within the cell to perform vital functions.
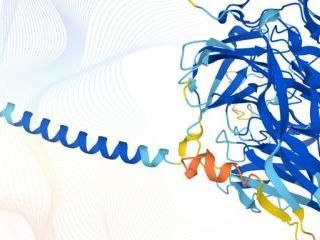
"Scientists know RNAPII's physical structure, but this large enzyme has many distinct regions that each perform distinct functions" said Dr. Kaplan, who is also a scientist at Texas A&M AgriLife. "We wanted to connect the dots between these regions and their function."
In laboratory experiments, the team took a genetic approach—generating 53 variations of RNAPII, so-called RNAPII "mutants," that each changed a specific part of RNAPII. They wanted to test each mutant against a particular function. In this way, they could link a specific region in the enzyme to a specific function.
But to do so, they had to compare each of the mutants against thousands of functions that RNAPII might be involved in within the cell—an immense task that couldn't be accomplished by traditional methods. So the team developed the pE-MAP approach.
"Instead of crossing a single point mutant with one or two different mutants, pE-MAP lets us cross each one with more than 1,000," said Dr. Krogan, who also directs the California Institute for Quantitative Biosciences, or QB3, at UCSF. "This gives us 1,000 data points for each mutant, which we then use to build our high-resolution profiles."
"Until now, the only way to get similar information was to deactivate, or "knock out" specific genes within an enzyme and observe the impact," explained Hannes Braberg, a graduate student in Dr. Krogan's lab and the paper's lead author. "But RNAPII is so critical that deactivating even one gene often kills the cell. So instead of knocking out the genes, we mutated them."
The team then correlated the newly generated profiles to how well each variation of RNAPII could transcribe DNA into RNA—the enzyme's most important function. The research team found that some of the RNAPII mutants transcribed more slowly than the others, while others were much faster. Further analysis revealed that the slow transcribers showed key similarities with each other, as did the fast transcribers. A pattern began to emerge that allowed them to predict the transcription speed—fast or slow—for each mutant.
And then the team discovered yet another phenomena related to transcription, involving a process called splicing, whereby specific stretches of non-coding RNA are cut out, and what remains is stitched back together. Previously, scientists had hypothesized that the transcription speed was related to splicing—in that fast transcribers would be less accurate splicers, and vice versa. But no one had been able to see it in action. So Dr. Guthrie, whose lab researches splicing, used the profiles generated from the pE-MAP approach to observe in real time how different transcription speeds affected splicing precision.
"When you slow down transcription, splicing gets more efficient," said Dr. Guthrie. "We saw the opposite effect in our fast transcribers—which had long been predicted but had never before been observed. This was another testament to the power of the pE-MAP approach."
The approach used here could be applied to studies of other enzymes, explained the authors. And what the researchers learn could then be used to develop an understanding of how mutations in enzymes like RNAPII lead to specific disease states—and may ultimately inform our ability to correct them.
Other news from the department science




















Get the life science industry in your inbox
From now on, don't miss a thing: Our newsletter for biotechnology, pharma and life sciences brings you up to date every Tuesday and Thursday. The latest industry news, product highlights and innovations - compact and easy to understand in your inbox. Researched by us so you don't have to.