Eine 3D-Karte des Genoms
Anzeigen



Meterlang ist der Erbgutfaden, der in jeder Zelle des Körpers steckt. Die Zellen müssen ihn sauber und ordentlich in einem Kern von nur fünf Mikrometern Durchmesser verstauen. Das funktioniert dank einer kunstvollen Falttechnik, durch die ein räumliches Zusammenspiel zwischen Genen und ihren Schaltern entsteht – mit Auswirkungen auf die menschliche Gesundheit. Ein internationales Team stellt nun in der Fachzeitschrift Nature eine neue Methode vor, mit der sich die dreidimensionale Topographie des gesamten Genoms kartieren lässt.
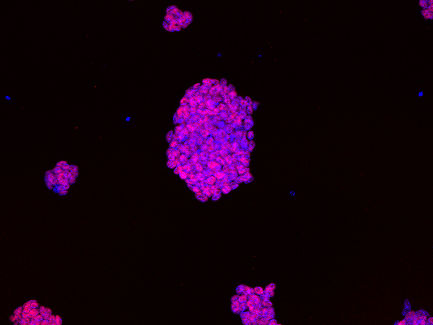
Kolonie embryonaler Stammzellen von einer Maus unter dem Mikroskop, die Zellkerne sind blau gefärbt. Die DNA der Zellkerne wird sequenziert, um die Positionen von Genen und Genschaltern zu bestimmen.
C. Ferrai, MDC
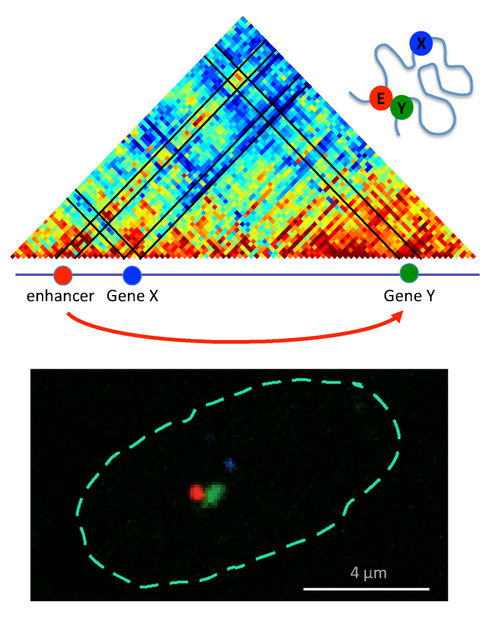
Lagebeziehungen zwischen Gen und Genschalter. Oben: Die Farben im Diagramm sind rötlicher, desto näher sich die Sequenzen sind. Der Genschalter (Enhancer, E) befindet sich nah an Gen Y, nicht Gen X.
AG Pombo, MDC
Wenn Gene aktiviert werden, werden sie zunächst in RNA und dann in Proteine übersetzt. Sobald diese Moleküle nicht mehr benötigt werden, wird das Gen wieder abgeschaltet. Dabei können sowohl die Gene als auch die dazugehörigen Schalter auf dem linearen Genom nah beieinander oder auch weit voneinander entfernt liegen. Die Zelle muss die beiden DNA-Abschnitte in Kontakt bringen, damit das Gen aktiviert werden kann.
Wer verstehen will, wie und wann Zellen ihre Gene an- und ausknipsen, muss also zu jedem Gen die jeweiligen Genschalter finden. Mit dem Mikroskop ist das nicht möglich, denn die DNA-Stränge sind zu fein, um ihre Bewegungen nachzuverfolgen. Zudem wäre die DNA-Menge im Zellkern zu groß. Man stelle sich ein riesiges Knäuel verhedderter Fäden vor, in dem man genau den Ort finden muss, an der sich zwei bestimmte Fäden treffen.

Kontakte finden – mit der Macht der Statistik und hauchfeinen Zellkernscheiben
Das „Genome Architecture Mapping“ (GAM) ist eine neue Methode, mit deren Hilfe Forscher diese Kontaktpunkte jetzt besser bestimmen können. Dabei gefrieren sie Gewebe oder Zellen zuerst und schneiden sie in hauchdünne Scheiben. Dann können sie die winzige DNA-Menge in den Schnitten einzelner Zellkerne sequenzieren. Dank eines Algorithmus namens SLICE können sie dann jene Regionen auf der DNA identifizieren, die bevorzugt miteinander interagieren. SLICE bestimmt die Häufigkeit, mit der verschiedene DNA-Abschnitte in einer Zellkernscheibe auftreten und schließt so auf die relative Position von Genen und den Regionen, die diese aktivieren.
„Wir wollen Beziehungen und Interaktionen verstehen. Eine Analogie wären Freundschaften zwischen Schülern einer Schule. Um dieses Rätsel zu lösen, könnten wir die Kinder zufällig im Speisesaal und auf dem Schulhof fotografieren“, erklärt Professor Ana Pombo, Leiterin der Studie. Sie begann mit der Arbeit an dem Forschungsprojekt am MRC London Institute of Medical Sciences (LMS) und arbeitet heute am Max-Delbrück-Centrum für Molekulare Medizin in der Helmholtz-Gemeinschaft (MDC) und dem Berliner Institut für Gesundheitsforschung (BIH). „Wenn wir über einen Monat hinweg viele solcher Fotos machen, würden wir irgendwann Muster erkennen: Welche Schüler sitzen häufig zusammen oder spielen gemeinsam? So verraten scheinbar willkürliche Schnappschüsse in ihrer Gesamtheit viel über die sozialen Interaktionen.“
Ähnlich funktioniert der SLICE-Algorithmus: „Mit mathematischen Methoden können wir Zufallsbegegnungen herausrechnen und sie von echten Interaktionen unterscheiden“, beschreibt der Professor Mario Nicodemi von der Universität Neapel, einer der leitenden Autoren, das Vorgehen. Nicodemi konzipierte die nötigen mathematischen Modelle und entwickelte sie gemeinsam mit seinem Doktoranden Antonio Scialdone.
Die grundlegende Idee zu dem Verfahren hatten Pombo und Dr. Paul Edwards vom Hutchison/MRC Research Centre and Department of Pathology der Universität Cambridge schon, bevor es die nötigen experimentellen Methoden überhaupt gab. „Mein Forschungsteam hat den Ansatz immer weiter verfeinert und neue technische Entwicklungen in die Methode integriert“, sagt Pombo.
Die Suche nach Genen mit Bedeutung für Krankheiten
In der in Nature veröffentlichten Studie wenden die Forscher die Methode bei embryonalen Stammzellen von Mäusen an. Sie wollen so die Gene untersuchen, deren Aktivität bei einigen schweren Krankheiten gestört ist und interessieren sich insbesondere dafür, welche Rolle weit entfernte Gen-Schalter dabei spielen. Die neue Studie liefert eine lange Liste neuer Kandidaten, die Wissenschaftler nun genauer untersuchen können.
Kontakte zwischen zwei Erbgutfäden konnten in früheren Arbeiten bereits identifiziert werden. Wie häufig und damit wichtig sie sind, blieb aber unklar, sagt Pombo: „Sie erkennen zwar, dass wir beide befreundet sind, aber nicht, wie eng die Freundschaft im Vergleich zu allen anderen ist.“
„Messungen zu Kontakten zwischen zwei DNA-Strängen gibt es schon lange“, bestätigt Dr. Robert Beagrie, einer der Erstautoren der Studie. Er erhob als Doktorand bei Pombo am LMS die Daten für die Studie und arbeitet nun an der Universität Oxford. „Diese Studien zeigen häufig, dass es Gruppen von DNA-Elementen gibt, die paarweise miteinander interagieren. Unser neuer Ansatz geht darüber hinaus. Er liefert einen genomweiten Katalog der Bereiche, die sogar gruppenweise interagieren.“ Damit kann das Forschungsteam auch Kontakte zwischen drei Erbgutsträngen verlässlich erkennen und quantifizieren. In diesen Abschnitten sind Gene besonders aktiv.
Großes Potenzial für die Untersuchung seltener Zelltypen
Die GAM-Methode arbeitet mit einzelnen Zellen und kann deren Position im Gewebe genau bestimmen. Sie kann auch solche Typen von Zellen untersuchen, die selten in einem Gewebe vorkommen – ein bedeutender wissenschaftlicher Fortschritt gegenüber früheren Methoden, die zahlreiche gleichartige Zellen brauchen und damit die Untersuchung solcher Zelltypen erschweren.
„Die Methode hat ungeheures Potenzial. Mit ihr können wir menschliche Gewebeproben untersuchen und die Kontakte zwischen den regulatorischen Bereichen und ihren entsprechenden Zielgenen katalogisieren. Diese Erkenntnisse lassen sich nutzen, um die genetische Variation und ihren Einfluss auf die Biologie des Zellkerns zu verstehen“, sagt Pombo.
Andere Forscher möchten mit der Methode nun untersuchen, was geschieht, wenn Retroviren ihre DNA in das Erbgut der Wirtszelle einschleusen. Auch für die Krebsforschung ist es interessant, die DNA von unterschiedlichen Bereichen eines Tumors zu kartieren. „Aus den GAM-Daten und mit Hilfe der mathematischen Modelle können wir diese Informationen verlässlich ableiten“, sagt Nicodemi. „So lassen sich jene Wechselwirkungen in Gruppen identifizieren, die eine Schlüsselrolle bei der Steuerung von Genen spielen.“
Beagrie fügt hinzu: „Wir stellen uns nun die Frage, ob ein Gen mit allen Genschaltern gleichzeitig in Kontakt kommt oder nur von jeweils einem zu einem gegebenen Zeitpunkt. Viele Gene, die für die frühe Entwicklung wichtig sind, besitzen mehrere dieser Schalter. Aber wie und warum diese zur Genregulierung aktiv werden, ist noch unklar.“
Originalveröffentlichung



Weitere News aus dem Ressort Wissenschaft


















































