Proteine bei der Arbeit beobachtet
Wissenschaftler bilden die aktivierte Strukur der Sensor-Histidinkinase nach
Anzeigen



Proteine steuern viele Prozesse im Körper, dabei verändern sie ihre Struktur. Die aktivierte Struktur ist experimentell jedoch schwer zugänglich. Über Genomanalyse, Computersimulationen und Laborexperimente hat ein internationales Forscherteam erstmals ein Modell der aktivierten Sensor-Histidinkinase, eines Proteins zur Signalübertragung, erarbeitet. Dr. Alexander Schug vom Karlsruher Institut für Technologie (KIT) bildete die Aktivierung in umfangreichen Computersimulationen nach. In der Zeitschrift PNAS stellen die Forscher ihre Ergebnisse vor, die Methode lässt sich auch auf andere Proteine übertragen.
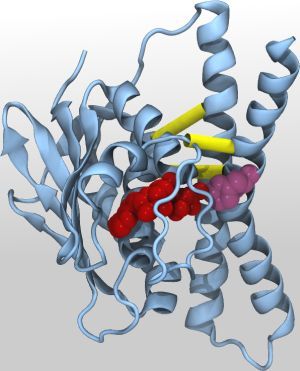
Aktivierte Anordung des Sensor-Histidinkinase-Proteins.
Dr. Alexander Schug
Während Proteine ihre Funktion erfüllen, verändern sie häufig ihre Struktur. Diese aktivierte Struktur ist oft nur kurzlebig und daher experimentell schwer zugänglich. Um die Funktion eines Proteins zu verstehen und eventuell gezielt zu beeinflussen, etwa bei der Behandlung von Krankheiten mit Medikamenten, ist es jedoch erforderlich, die Strukturveränderungen genau zu kennen.
In einem Projekt, das Genomanalyse, Computersimulation und Experimente zur Erbgutveränderung verbindet, haben Forscher aus den USA, Frankreich und Deutschland ein Strukturmodell einer schwer fassbaren aktivierten Anordnung eines wichtigen Proteins erarbeitet. Die Computersimulation und Strukturnachbildung übernahm Dr. Alexander Schug, Leiter der Helmholtz Junior Research Group „Multiscale Biomolecular Simulation“ am Steinbuch Centre for Computing (SCC) des KIT.
Die Wissenschaftler konzentrierten sich auf Zwei-Komponenten-Signalübertragungssysteme, die besonders in Bakterien sehr häufig sind. Solche Systeme bestehen aus einem Sensor-Histidinkinase-Protein als Empfänger für Signale von außen, das die Informationsübertragung durch eine sogenannte Autophosphorylierung einleitet, und einem Antwortregulator-Protein. Über diese Systeme lagen bisher nur teilweise Strukturinformationen vor.
Durch eine statistische Analyse einer großen Menge von Genomdaten identifierten die Forscher Teile des Sensor-Histidinkinase-Proteins, die während der Stukturveränderungen miteinander in Kontakt treten oder den Kontakt zueinander abbrechen. Basierend auf dieser Analyse, gelang es dem KIT-Forscher Dr. Alexander Schug, in umfangreichen Computersimulationen die Strukturveränderungen während der Autophosphorylierung nachzubilden und ein Modell der aktivierten Struktur zu erstellen. Dieses Modell ließ sich anschließend in Laborexperimenten verifizieren.
Zwei-Komponenten-Systeme stellen bei allen Bakterien das primäre Signal-Reaktions-System dar. Daher tragen die Ergebnisse der Forscher zum Verständnis der bakteriellen Signalübertragung bei. Die Erkenntnisse könnten künftig die Entwicklung neuer Antibiotika voranbringen. Überdies ist der Ansatz auch für andere Proteinsysteme relevant: „Da die reine Menge an Genomdaten in den vergangenen zehn Jahren geradezu explodiert ist und weiter rasant wächst, lässt sich unsere Methode auf immer mehr Proteine übertragen, auch über die Signalübertragung hinaus“, erklärt Schug.
Originalveröffentlichung
Weitere News aus dem Ressort Wissenschaft