Krankheitsgene können die Hirnentwicklung retten
Angeborene Erkrankungen entschlüsseln und vielleicht verhindern
Wenn sich beim ungeborenen Kind die vorderen Hirnhälften nicht oder unvollständig teilen, entsteht eine Holoprosenzephalie. Das MDC-Team um Annette Hammes stellt nun im Fachjournal „Development“ Kandidatengene vor, die den Schweregrad dieser angeborenen Fehlbildung des Vorderhirns positiv beeinflussen können.
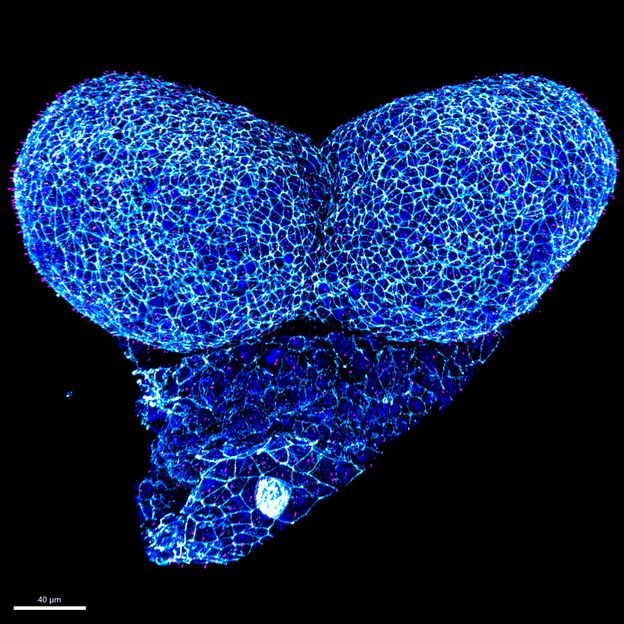
Vordere Neuralfalten in einem Mausembryo.
Hammes Lab, MDC
Bei etwa einem bis vier von 1.000 Ungeborenen kommt es zu einer Holoprosenzephalie (HPE), bei der sich die vorderen Hirnhälften nicht oder nur unvollständig teilen. Mit dieser häufigsten Fehlbildung des Vorderhirns beim Menschen gehen Entstellungen des Gesichts einher, etwa Lippen-Kiefer-Gaumen-Spalten oder sehr nah beieinanderstehende Augen bis hin zur Verschmelzung der beiden Augäpfel. Die meisten Föten sterben noch im Mutterleib.
Die Ursachen der HPE sind noch nicht genau geklärt. Neben Umweltgiften und Erkrankungen der werdenden Mutter spielen genetische Faktoren eine Rolle, darunter Mutationen in Genen des Sonic-Hedgehog (SHH)-Signalwegs. Dieser Signalweg steuert die Entwicklung der Organe und des Nervensystems während der Entwicklung des Embryos. Gendefekte und ein dadurch bedingter Funktionsverlust von Lrp2, einem SHH-Co-Rezeptor, führen zu sehr unterschiedlichen Ausprägungen der Hirndefekte. „Wir wollten wissen, warum der Schweregrad dieser Erkrankung so stark variiert“, sagt Dr. Annette Hammes, die am Max-Delbrück-Centrum für Molekulare Medizin in der Helmholtz-Gemeinschaft (MDC) die Arbeitsgruppe „Molekulare Signalwege in der kortikalen Entwicklung“ leitet. „Während manche Betroffene keine oder milde Symptome haben, müssen andere mit schweren Missbildungen leben – selbst wenn sie miteinander verwandt sind, also davon auszugehen ist, dass die Ursache für die Erkrankung dieselbe Genmutation ist.“

Krankheitsgene restaurieren den SHH-Signalweg
Forscher gehen seit längerem davon aus, dass es Gene gibt, die die Fehlbildung positiv beeinflussen oder sogar ganz verhindern. Das Team der AG Hammes hat nun zwei neue Kandidaten identifiziert. „Es handelt sich um Ulk4 und Pttg1, auch als Securin bekannt“, sagt Erstautorin Dr. Nora Mecklenburg, die zum Zeitpunkt der Studie Postdoktorandin bei Annette Hammes war. ULK4 ist ein Gen, mit dem bislang Schizophrenie und bipolare Störungen assoziiert sind; PTTG1 wird hauptsächlich im Zusammenhang mit Krebserkrankungen erforscht. Im Fachjournal „Development“ beschreiben die Wissenschaftler, dass diese Proteine einen gestörten SHH-Signalweg restaurieren können. Neun Jahre Forschung stecken in der Studie, die das Journal als „Research Highlight“ bezeichnet.
Ihre Ergebnisse haben sie zum Teil auch dem Zufall zu verdanken. Zusammen mit der MDC-Forschungsgruppe um Professor Thomas Willnow hatte Annette Hammes mit ihrem Team über viele Jahre hinweg Mäuse mit Lrp2-Mutation untersucht. „Wir wissen, dass Lrp2 früh in der Embryogenese die Entstehung des Neuralrohrs beeinflusst, aus dem sich später das Nervensystem entwickelt“, sagt die Neurowissenschaftlerin. Ohne Lrp2 wird der SHH-Signalweg nicht ausreichend aktiviert, und es kommt in einer sehr frühen Phase der Schwangerschaft zu Fehlbildungen des Neuralrohrs, die sehr häufig zu einer Fehlgeburt führen. Als die Wissenschaftler die Lrp2-Mutanten des üblicherweise verwendeten Mausstammes mit schwarzer Fellfarbe, kurz „Black6“-Mäuse genannt, in einen anderen Mausstamm mit weißer Fellfarbe einkreuzten, war die Überraschung groß: Die Nachfahren mit weißem Fell wiesen keinerlei Fehlbildungen des Gehirns oder im Gesicht auf – trotz Mutation im SHH-Co-Rezeptor Lrp2. Sie schlussfolgerten, dass es bisher unbekannte Faktoren geben muss, die den SHH-Signalweg beeinflussen, und machten sich auf die Suche.
RNA-Analyse im Hochdurchsatzverfahren
Dafür züchtete Nora Mecklenburg zunächst die verschiedenen Mausstämme und untersuchte die Tiere hinsichtlich ihrer Krankheitsmerkmale, der Signalwege und ihrer Genetik. Zusammen mit ihren Co-Erstautorinnen Franziska Witte, damals Doktorandin in der MDC-Arbeitsgruppe von Professor Norbert Hübner, und Izabela Kowalczyk, Doktorandin bei Annette Hammes, sequenzierte und analysierte sie im Hochdurchsatzverfahren die RNA embryonaler Zellen der verschiedenen Stämme. Dabei stellten die drei Wissenschaftlerinnen fest, dass die Zellen trotz einer ähnlichen Genomsequenz völlig unterschiedliche Transkriptome aufweisen, die Gene also sehr unterschiedlich abgelesen werden. „Das Ausmaß der Unterschiede selbst zwischen den Wildtypen der beiden Mausstämme zu diesem frühen Zeitpunkt der Embryonalentwicklung hat uns wirklich überrascht“, sagt Nora Mecklenburg.
In weiteren Untersuchungen fanden die Forscher heraus, dass in den weißen Lrp2-Mutanten wie auch bei den Wildtypen dieses Stammes einige Gene, darunter Ulk4 und Pttg1, im Vergleich zu den Black6-Tieren stark hochreguliert sind. Um zu sehen, ob sich das auf den SHH-Signalweg auswirkt, brachten sie die Gene in Zellen ohne Lrp2-Funktion ein. „Wir konnten beobachten, dass sie den Sonic-Hedgehog-Signalweg deutlich ankurbeln“, erzählt Izabela Kowalczyk. Ihre Schlussfolgerung: „Ulk4 und Pttg1 werden in den Lrp2-Mutanten mit den weißen Vorfahren vermehrt gebildet. Sie kompensieren das fehlende Lrp2 und stellen einen ausreichend starken SHH-Signalweg wieder her. Die Fehlbildungen bleiben aus.“ Diese Erkenntnis lässt die Krankheitsgene Ulk4 und Pttg1 in einem völlig neuen Licht erscheinen: Während sie im erwachsenen Organismus bei erhöhter Expression Krankheiten auslösen können, können sie die Entwicklung eines Embryos positiv beeinflussen. Die Wissenschaftler konnten auch den Ort ausfindig machen, von dem aus diese Faktoren den SHH-Signalweg verstärken – in den Antennen-ähnlichen Fortsätzen der Neuroepithelzellen, den Zellen, die das Neuralrohr von innen auskleiden.
Angeborene Erkrankungen entschlüsseln und vielleicht verhindern
„Dass wir diese Kandidatengene, die den SHH-Signalweg modulieren, in der Maus identifiziert haben, bringt unser Wissen zur Holoprosenzephalie und anderer angeborener Erkrankungen einen Schritt weiter“, sagt Annette Hammes. „So finden wir vielleicht eine Möglichkeit, sie zu verhindern.“ Doch bis dahin ist es noch ein langer Weg – ein Weg, auf dem ihr Team als nächstes erforschen möchte, welche Rolle die neu entdeckten SHH-Signalweg-Modifikatoren nicht nur während der embryonalen Entwicklung, sondern auch im erwachsenen Gehirn spielen. So haben die Wissenschaftler PTTG1 bereits im Zytoskelett von Neuronen ausfindig gemacht – einem Protein-Netzwerk im Zytoplasma, das den Zellen Stabilität verleiht. Das Team untersucht derzeit, welche Funktion das Gen dort hat.
Originalveröffentlichung
Weitere News aus dem Ressort Wissenschaft





















































