Zielgerichteter Therapieansatz für seltene Knochenkrebsart
Chordome sind seltene Knochentumoren, die nur schlecht behandelt werden können. Wissenschaftler und Ärzte vom Nationalen Centrum für Tumorerkrankungen (NCT), Deutschen Krebsforschungszentrum (DKFZ) und Universitätsklinikum Heidelberg (UKHD) konnten mittels einer Genanalyse ein besonderes genetisches Merkmal von Chordomen im fortgeschrittenen Stadium aufdecken. Ihre im Fachblatt Nature Communications veröffentlichten Ergebnisse deuten darauf hin, dass eine Gruppe von Arzneistoffen, die bereits bei der Behandlung anderer Krebsarten zugelassen ist, auch gegen Chordome wirksam sein könnte.
Nationales Centrum für Tumorerkrankungen (NCT) Heidelberg, eine gemeinsame Einrichtung des Deutschen Krebsforschungszentrums (DKFZ), des Universitätsklinikums Heidelberg (UKHD) und der Deutschen Krebshilfe.
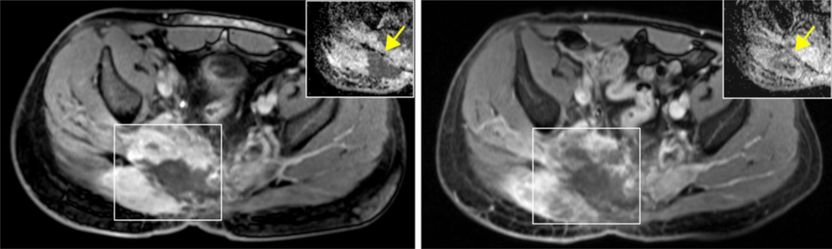
links: Patient mit fortgeschrittenem Chordom; rechts: Stillstand des Tumorwachstums nach fünf Monaten Behandlung mit PARP-Inhibitor
NCT Heidelberg
Chordome sind sehr seltene Tumoren der Wirbelsäule. Sie machen ungefähr ein Prozent aller Knochentumoren aus. Sie zählen zwar zur Klasse der Knochentumoren, entstehen allerdings nicht aus Knochengewebe, sondern aus Resten der so genannten Chorda dorsalis. Bei allen Wirbeltieren wird die Chorda dorsalis embryonal angelegt und im Laufe der Embryonalentwicklung bis auf Rudimente von der Wirbelsäule ersetzt. Üblicherweise treten Chordome jenseits des 30. Lebensjahres auf, wobei Männer und Frauen gleich häufig von dem Wirbelsäulentumor betroffen sind. Die Behandlung gestaltet sich oft schwierig, da Chordome in der Regel gegen eine konventionelle Chemotherapie resistent sind. Ärzte versuchen, das betroffene Gewebe chirurgisch zu entfernen, was allerdings oft nicht vollständig gelingt. Die Mehrheit der Betroffenen wird daher anschließend mit Strahlentherapie behandelt. In rund zwei Dritteln der Fälle kommt der Tumor jedoch zurück. Forscher suchen deshalb nach neuen Wegen und Ansätzen, um dieser Erkrankung beizukommen.
Wissenschaftler und Ärzte vom NCT Heidelberg, UKHD und DKFZ haben nun in Zusammenarbeit mit Kollegen der Standorte des Deutschen Konsortiums für Translationale Krebsforschung (DKTK) detaillierte Genanalysen der Tumorzellen von Chordom-Patienten durchgeführt. Die Arbeit fand im Rahmen des NCT/DKTK MASTER-(Molecularly Aided Stratification for Tumor Eradication)-Programms statt. Die Studie unter Leitung des Kommissarischen Geschäftsführenden Direktors am NCT Heidelberg Stefan Fröhling richtet sich vor allem an junge Patienten mit fortgeschrittenen Krebserkrankungen und Patienten mit sehr seltenen Tumoren.
Insgesamt untersuchte das Team elf Chordom-Patienten im fortgeschrittenen Stadium, bei denen die Standardtherapien bereits ausgeschöpft waren. Die Wissenschaftler sequenzierten das Erbgut der Krebszellen vollständig und entdeckten, dass fortgeschrittene Chordome bestimmte molekulare Veränderungen aufweisen, die mit einer gestörten DNA-Reparatur durch die so genannte homologe Rekombination (HR) verbunden sind.
Im Allgemeinen verwenden Zellen die HR, um schadhafte Stellen der DNA-Stränge zu reparieren. Es ist bekannt, dass auch bei anderen Krebszellarten die HR beeinträchtigt ist. Die Diagnose dieser fehlerhaften Funktion erfordert bestimmte Voraussetzungen. "Allerdings trafen nur bei drei der elf untersuchten Patienten diese klassischen Kriterien zu", berichtet Stefan Gröschel, Oberarzt am UKHD und Leiter der Arbeitsgruppe Molekulare Leukämogenese am DKFZ. "Bei Chordomen scheinen also offenbar weitere, noch unbekannte genetische Veränderungen zu einer Beeinträchtigung der HR zu führen."
Da sich bei anderen Krebsarten, bei denen ebenfalls ein HR-Defizit vorliegt, bestimmte Arzneistoffe als wirksame Medikamente erwiesen haben, lag es nahe, diese auch bei Chordom-Patienten als weitere Behandlungsoption einzusetzen. Die Ärzte führten bei einem Betroffenen mit passendem genetischem Profil eine experimentelle Behandlung mit einem so genannten PARP-Inhibitor durch. PARP-Inhibitoren hemmen das Enzym Poly-ADP-Ribose-Polymerase (PARP) und verhindern dadurch, dass Krebszellen Schäden an ihrer DNA, die etwa in Folge einer Chemotherapie auftreten, wieder reparieren können. Bei dem behandelten Patienten führte die Gabe eines PARP-Inhibitors zu einer langanhaltenden klinischen Verbesserung und einem Stillstand des Tumorwachstums. Nach erneutem Fortschreiten der Erkrankung bei demselben Patienten konnte das Team um Stefan Fröhling, Stefan Gröschel und Robert Russell vom BioQuant Heidelberg eine neuartige Resistenzmutation des PARP1-Enzyms identifizieren, welche die Wirkung des PARP-Inhibitors aufhob.
"Unsere Ergebnisse zeigen, wie die Suche nach neuen personalisierten Krebstherapien im klinischen Alltag funktionieren kann. Durch den Einsatz eines zugelassenen Medikaments, das bisher bei Chordomen noch nicht angewendet wurde, konnten wir für einen Patienten über einen Zeitraum von zehn Monaten eine verbesserte Erkrankungssituation erreichen. Und auch wenn die Erkrankung danach erneut fortgeschritten ist, hoffen wir, dass uns der neu entdeckte Resistenzmechanismus zukünftig helfen wird, Therapien besser zu planen und früher auf Veränderungen in der Wirksamkeit der Medikamente reagieren zu können", berichtet Fröhling.
Originalveröffentlichung
Weitere News aus dem Ressort Wissenschaft



















































