Wie ein mutiertes Gen Bluthochdruck hervorrufen kann
Eine der häufigsten Ursachen von sekundärem Bluthochdruck, auch arterielle Hypertonie genannt, ist der primäre Hyperaldosteronismus, kurz PA. Dabei produziert die Nebennierenrinde zu große Mengen des Hormons Aldosteron, das eine wichtige Rolle in der Regulierung des Salz- und Wasserhaushalts und des Blutdrucks spielt. Hohe Konzentrationen von Aldosteron bewirken, dass in der Niere vermehrt Natrium und Wasser rückresorbiert werden. Dadurch erhöht sich das Blutvolumen und der Blutdruck steigt an.
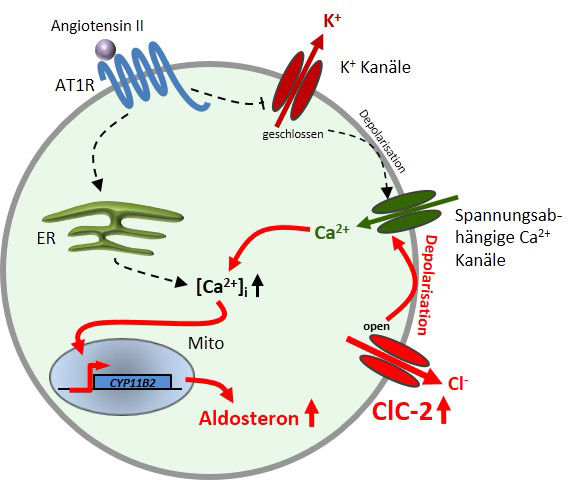
Eine Mutation im Chloridkanal ClC-2 als Ursache für primären Hyperaldosteronismus (PA). Die Bildung des an der Blutdruckregulation maßgeblich beteiligten Hormons Aldosteron ist im gesunden Menschen strikt reguliert: Sobald Angiotensin II an seinen Rezeptor bindet, werden K+-Kanäle geschlossen und die Zelle wird depolarisiert. Die darauffolgende Öffnung spannungsabhängiger Ca2+-Kanäle bewirkt eine Erhöhung intrazellulären Ca2+, was über eine Signalkaskade die Bildung von Aldosteron induziert. In dieser Studie wurde eine Patienten-Mutation im Chloridkanal ClC-2 charakterisiert, die eine Depolarisation durch den Ausstrom von Cl- durch den ständig geöffneten Kanal hervorruft. Die Folge dieser Mutation ist eine Angiotensin II-unabhängige Überproduktion von Aldosteron, die zu Bluthochdruck führt.
Der PA wird durch eine Genveränderung ausgelöst, die dazu führt, dass in den Zellen der Nebennierenrinde ein Ionenkanal namens ClC-2 permanent offen steht. Das hat ein Team um Prof. Dr. Thomas Jentsch vom Leibniz-Forschungsinstitut für Molekulare Pharmakologie (FMP) und vom Max-Delbrück-Centrum für Molekulare Medizin in der Helmholtz-Gemeinschaft (MDC) in Berlin in Zusammenarbeit mit Pariser Kollegen um Dr. Maria Christina Zennaro am INSERM, ein international bekanntes Team im Feld des PA, jetzt herausgefunden. Hierdurch kommt es zu einer Signalkaskade, die letztlich eine vermehrte Produktion von Aldosteron zur Folge hat.
Das Exom von Patienten verriet, warum zu viel Aldosteron produziert wird
„Bekannt waren bisher nur Knock-out-Mutationen des CLCN2-Gens“, erläutert Jentsch. „Diese führen dazu, dass der ClC-2-Kanal gar nicht oder nur fehlerhaft gebildet wird.“ Das daraus resultierende Krankheitsbild ist unter anderem eine Leukodystrophie, bei der die weiße Substanz des Gehirns geschädigt wird. Symptome der Erkrankung sind beispielsweise Gangstörungen. „Die Genveränderung, auf die wir jetzt gestoßen sind, führt hingegen nicht zu einem Verlust, sondern umgekehrt zu einer Verstärkung der Ströme durch den Kanal“, sagt Jentsch. „Sie hat zur Folge, dass sich der Kanal nicht mehr wie gewöhnlich spannungs- und pH-abhängig schließt.“
Um den Ursachen des PA auf den Grund zu gehen, hatten die französischen Wissenschaftler das Exom – also all jene Abschnitte des Genoms, die Bauanleitungen für Proteine enthalten – von Patienten analysiert, bei denen die Krankheit schon vor dem 25. Lebensjahr ausgebrochen war, und es mit dem Exom gesunder Menschen verglichen. Dabei stießen sie auf eine noch unbekannte Mutation im CLCN2-Gen und wandten sich an Jentsch, den weltweit führenden Experten für CLC Kanäle.
„Wir haben daraufhin eine Hypothese entwickelt, wie die Mutation zu der Krankheit PA führen könnte“, berichtet Corinna Göppner aus der Arbeitsgruppe von Thomas Jentsch, die an der Studie beteiligt war. Normalerweise führt in den Zellen der Nebennierenrinde das Hormon Angiotensin II dazu, dass Kaliumkanäle geschlossen werden und keine positiv geladenen Kaliumionen mehr ausströmen können. Dadurch verändert sich das Membranpotential, es kommt zur Depolarisation und in Folge dessen zu einer Öffnung spannungsabhängiger Kalziumkanäle. Daraufhin strömen Kalziumionen in die Zelle ein und setzen eine Signalkaskade in Gang, an deren Ende die Produktion von Aldosteron steht.
Bei Mäusen existiert der Kanal in der Nebennierenrinde
„Faszinierenderweise lag die Patientenmutation genau in einem von Jentsch schon 1992 identifizierten Abschnitt von ClC-2, in dem Mutationen den Kanal öffnen. Wir vermuteten nun, dass ein offener Chloridkanal aufgrund des permanenten Ausstroms von negativ geladenem Chlorid die Spannung der Zellen ändert und dadurch die Kalziumkanäle unabhängig von Angiotensin II öffnet“, sagt Göppner. „In diesem Fall würde Aldosteron fortlaufend von den Zellen gebildet und freigesetzt.“
Im Mausmodell konnten die Forscher zunächst zeigen, dass der ClC-2-Kanal in den Zellen der äußeren Schicht der Nebennierenrinde, der Zona glomerulosa, tatsächlich existiert. „Per Patch-Clamp-Technik ließ sich nachweisen, dass Chloridionen durch diesen Kanal hindurchfließen“, erläutert Dr. Ian Orozco, der einen Großteil der Experimente durchgeführt hat. „Der Stromfluss blieb jedoch aus, wenn in den Mäusen das Gen für den Kanal stillgelegt war.“
In einem nächsten Schritt verglichen die Forscher den Kanal mit seinem mutierten Pendant nach Produktion in Eizellen von Krallenfröschen. „Hier zeigte sich, dass die in den Patienten gefundene Genveränderung in der Tat zu einem verstärkten Stromfluss führt und sich der ClC-2-Kanal nicht mehr wie gewohnt regulieren lässt“, sagt Orozco.
Welche Substanz kann den Erkrankten helfen?
In Zelllinien der humanen Nebennierenrinde konnten das französische Team nachweisen, dass die Zellen unter der Mutation tatsächlich eine geringere Membranspannung aufweisen und mehr Aldosteron produzieren. Auch die Enzyme, die an der Herstellung des Hormons beteiligt sind, wurden in den genveränderten Zellen vermehrt gebildet. Mit Substanzen, die die Kalziumkanäle blockieren, ließ sich die Überproduktion hingegen stoppen. „Damit war unsere eingangs aufgestellte Hypothese sehr gut belegt“, sagt Göppner.
Denkbar wäre nun die Entwicklung einer Substanz, die spezifisch den ClC-2-Kanal in den Zellen der Nebennierenrinde blockiert, um Patienten mit dieser Form von PHA zu heilen. Das allerdings ist noch Zukunftsmusik.
Originalveröffentlichung
L. Fernandes-Rosa, Georgios Daniil, Ian J. Orozco, Corinna Göppner, Rami El Zein, Vandana Jain, Sheerazed Boulkroun, Xavier Jeunemaitre, Laurence Amar, Hervé Lefebvre, Thomas Schwarzmayr, Tim M. Strom, Thomas J. Jentsch and Maria-Christina Zennaro; "A gain-of-function mutation in the CLCN2 chloride channel gene causes primary aldosteronism"; Nature Genetics; 2018
Weitere News aus dem Ressort Wissenschaft




















































