UDE: Einblicke in die Entstehung von Knochentumoren
Einer seltenen Erkrankung des Skeletts sind Wissenschaftler der Uni Duisburg-Essen (UDE) auf der Spur. Sie verursacht bei den Betroffenen - besonders bei Kindern - oft Bewegungseinschränkungen, Schmerzen und ein verkürztes Wachstum der Knochen. Dabei entstehen in der Nähe der Gelenke gutartige Knochentumoren, sogenannte kartilaginäre Exostosen oder Osteochondromen. Anhand eines neuen Mausmodells lassen sich diese nun bereits in einem frühen Stadium erforschen.
Das Modell haben Forscher des Zentrums für Medizinische Biotechnologie (ZMB) an der UDE mit Kooperationspartnern vom Huntsman Cancer Institut der Universität Utah in den USA entwickelt. Sein Krankheitsbild ähnelt dem menschlichen Phänotyp stark. "Mithilfe dieses Mausmodells ist es erstmals möglich, die Entstehung der Tumoren in den frühesten Anfangsstadien zu untersuchen und Einblicke in die molekularen Ursachen zu gewinnen", so Prof. Dr. Andrea Vortkamp von der Abteilung Entwicklungsbiologie des ZMB. "Die neuen Erkenntnisse leisten zudem einen wichtigen Beitrag zu den Kontroversen über den zellulären Ursprung der Erkrankung - dieser wird seit vielen Jahren stark diskutiert", erklärt Vortkamp.
Die ersten Tumoren treten bei kleinen Kindern auf, wachsen unterschiedlich stark und verknöchern häufig. Selten entwickeln sich die gutartigen Geschwülste zu karzinogenen Knorpeltumoren (Chondrosarkome). In den meisten Fällen können sie auf einen genetischen Defekt in einem von zwei Genen, Ext1 oder Ext2, zurückgeführt werden. Die Patienten sind für die Mutation eines dieser Gene heterozygote Träger, das heißt, dass sie im Gegensatz zu gesunden Menschen nur über ein normales Allel und somit nur über die Hälfte der jeweiligen Genaktivität verfügen.
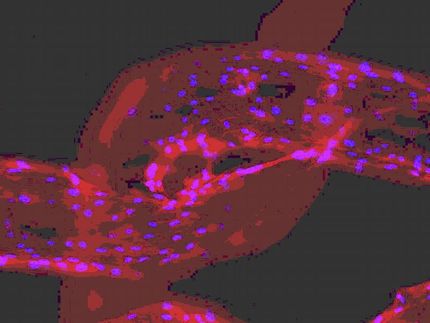
Bereits vor mehr als zehn Jahren haben Wissenschaftler ein erstes Mausmodell etabliert, um die Bildung der Knochentumoren auf molekularer und zellulärer Ebene untersuchen zu können - allerdings vergeblich: Die heterozygoten Ext1-Mäuse zeigen erstaunlicherweise kein dem Menschen vergleichbares Krankheitsbild.
Schon früh haben die Forscher deshalb vermutet, dass die Ausbildung der Tumore im Menschen durch eine zusätzliche Mutation des zweiten Allels verursacht wird (Verlust der Heterozygosität). Um diese Hypothese überprüfen und die Entstehung im Detail erforschen zu können, haben sie bei dieser Studie tief in die Trickkiste der Molekularbiologie gegriffen: Mithilfe eines DNA-Konstrukts, das in das Genom einer Maus eingeschleust wurde, konnten die Wissenschaftler das Ext1-Gen zu verschiedenen Zeitpunkten und in spezifischen Geweben ausschalten. So gab es in vereinzelten Zellen des ansonsten normalen Knorpelgewebes keine Ext1-Genaktivität mehr.
Auf diese Weise gelang es den Wissenschaftlern ein Mausmodell zu etablieren, welches zu 100 Prozent Osteochondrome ausbildet. Gleichzeitig konnten sie zwei lange umstrittene Fragen aufklären: Zum einen wurde gezeigt, welcher Zelltyp die Vorläuferzelle des Tumors bildet, zum anderen wurde die Tumor-auslösende genetische Konstellation festgestellt.
Dies sind wichtige Voraussetzungen, um künftig therapeutische und pharmakologische Strategien entwickeln zu können. Denn bis heute gibt es keine präventiv wirksamen Therapieansätze; die auftretenden Tumoren müssen oft mehrmals im Leben des Patienten operativ entfernt werden.
Ihre Erkenntnisse haben die Wissenschaftler in der Fachzeitschrift PNAS (Proceedings of the National Academy of Sciences of the United States of America) veröffentlicht.
Weitere News aus dem Ressort Wissenschaft

















































