Los científicos producen los primeros modelos de código abierto de la proteína COVID-19 'S' de longitud completa.
La estructura de la proteína facilita la entrada del virus en las células huésped, convirtiéndolo en un objetivo clave para el desarrollo de vacunas y medicamentos antivirales.
Anuncios



El virus SARS coronavirus 2 (SARS-CoV-2) es la causa conocida de la enfermedad coronavirus 2019 (COVID-19). El "pico" o la proteína S facilita la entrada del virus en las células huésped.
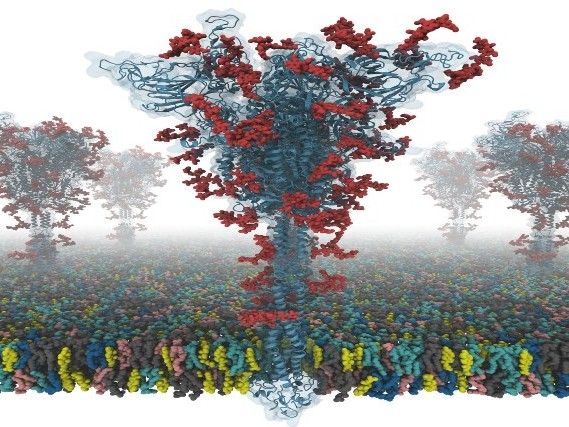
Un modelo de una proteína S.
Dr. Yeolkyo Choi/Lehigh
Ahora un grupo de investigadores de la Universidad Nacional de Seúl en Corea del Sur, la Universidad de Cambridge en el Reino Unido y la Universidad de Lehigh en EE.UU., han trabajado juntos para producir los primeros modelos de código abierto de todos los átomos de una proteína S de longitud completa. Los investigadores dicen que esto es de particular importancia porque la proteína S juega un papel central en la entrada del virus en las células, lo que la convierte en un objetivo principal para el desarrollo de vacunas y medicamentos antivirales.
Los detalles se pueden encontrar en el artículo "Developing a Fully-glycosylated Full-length SARS-CoV-2 Spike Protein Model in a Viral Membrane" que acaba de ser publicado en línea en The Journal of Physical Chemistry B.
Un video demostrativo ilustra cómo construir este sistema de membranas a partir de sus modelos de la proteína S del SARS-CoV-2. El programa de construcción de modelos es de acceso abierto y se puede encontrar en la página principal de CHARMM-GUI haciendo clic en el enlace de archivo de COVID-19.
Desarrollado por Wonpil Im, profesor del Departamento de Ciencias Biológicas y del Departamento de Bioingeniería de la Universidad de Lehigh, CHARMM-GUI (GUI = graphical user interface) es un programa que simula sistemas biomoleculares complejos de forma simple, precisa y rápida. Im lo describe como un "microscopio computacional" que permite a los científicos comprender las interacciones a nivel molecular que no pueden ser observadas de otra manera.
"Nuestros modelos son los primeros modelos de proteína de punta (S) de SARS-CoV-2 totalmente glicosilada que están disponibles para otros científicos", dice Im. "Tuve la suerte de colaborar con el Dr. Chaok Seok de la Universidad Nacional de Seúl en Corea y el Dr. Tristan Croll de la Universidad de Cambridge en el Reino Unido. Nuestro equipo pasó días y noches construyendo estos modelos muy cuidadosamente a partir de las porciones de estructura criogénica conocidas. El modelado fue muy difícil porque había muchas regiones donde el simple modelado no proporcionaba modelos de alta calidad".
Los científicos pueden utilizar los modelos para llevar a cabo una investigación de simulación innovadora y novedosa para la prevención y el tratamiento de COVID-19, según el Im.
La estructura de la proteína S se determinó con crio-EM con el RBD arriba (PDB ID: 6VSB), y con el RBD abajo (PDB ID: 6VXX). Pero, este modelo tiene muchos residuos que faltan. Así que, primero modelaron los residuos de aminoácidos faltantes, y luego otros dominios faltantes. Además, modelaron todos los glicanos potenciales (o carbohidratos) unidos a la proteína S. Estos glicanos impiden el reconocimiento de anticuerpos, lo que dificulta el desarrollo de una vacuna. También construyeron un sistema de membrana viral de una proteína S para la simulación de la dinámica molecular.
Nota: Este artículo ha sido traducido utilizando un sistema informático sin intervención humana. LUMITOS ofrece estas traducciones automáticas para presentar una gama más amplia de noticias de actualidad. Como este artículo ha sido traducido con traducción automática, es posible que contenga errores de vocabulario, sintaxis o gramática. El artículo original en Inglés se puede encontrar aquí.
Publicación original

Anuncios



Más noticias del departamento ciencias



















Noticias más leídas









Más noticias de nuestros otros portales








Contenido visto recientemente

Utilizar CRISPR para descifrar si las variantes genéticas provocan cáncer - Hay miles de mutaciones en el genoma humano de las que se sospecha que desempeñan un papel en el cáncer, pero cuya función exacta nunca se ha aclarado

Veneno de araña para la terapéutica y los bioinsecticidas - Nuevas biomoléculas a partir del veneno de la avispa

Variantes en los genes BRCA1/2 y MMR en niños con cáncer - Las pruebas genéticas no se recomiendan en niños sanos
Lesión cerebral traumática: la molécula de diseño apoya la reparación del cerebro - Un equipo internacional de investigación ha descubierto cómo se puede mejorar la reparación después de una lesión cerebral influyendo en las células inmunológicas
