Zooming in on genome breakages could reveal hidden secrets of cancer
Scientists at IFOM in Milan have uncovered the mechanisms that cause tumours to develop in patients suffering from Ataxia-Telangiectasia (AT) and Ataxia-Telangiectasia Like Disorder (ATLD): rare hereditary diseases that predispose afflicted individuals to cancer. The study opens new avenues for the development of personalized anti-cancer therapies and early diagnostic tests. The research is featured in Cell.
The cells in our body are continuously subjected to DNA damage. Normally, our genome is safeguarded against such damage thanks to specific genes that can detect DNA breaks and activate repair mechanisms. ATM is one of these key genes and its correct functioning is essential for genome stability. Since ATM also regulates p53, acknowledged as the most important tumour suppressor gene, it is not surprising that the lack of ATM dramatically increases the risk of developing tumours.
ATM is best known for its involvement in a rare hereditary disease characterized by the degeneration of a part of the brain and premature aging: Ataxia-Telangiectasia (AT), from which the name ATM (Ataxia Telangiectasia Mutated) is derived. In individuals suffering from this syndrome, mutations in the ATM gene prevent it from activating cell cycle control systems (or checkpoints), thus compromising the mechanisms that normally oversee repair of damaged DNA. Hence, in AT patients the probability of developing cancer is much higher (around 1000 times higher) than in normal individuals.
Patients who are homozygous for ATM mutations manifest the complete clinical symptoms of the disease. In contrast, individuals who are heterozygous for ATM mutations behave like “healthy carriers” and do not manifest any outward symptoms. However, these individuals are still predisposed to cancer and it has been estimated that they may represent as many as 1% of the population. Indeed, it has been shown that 9% of breast cancers occur in individuals heterozygous for AT. However, until now, it was not clear how ATM mutations predispose individuals to cancer and therefore how is best to prevent and treat cancer in these patients.
The study directed by Marco Foiani, Scientific Director of IFOM (FIRC Institute of Molecular Oncology) and Full Professor of Molecular Biology at the University of Milan (Department of Biomolecular Sciences and Biotechnology), analyzes in detail how cells respond to the absence of the repair proteins normally activated by the ATM gene. “When ATM, or the proteins that are regulated by it, do not work, – explains Foiani – DNA breaks can no longer be repaired. This leads to the accumulation of DNA damage, which can eventually trigger the onset of cancer”.
The scientists arrived at this conclusion using a particular experimental approach: “we are able to induce a single DNA breakpoint at an exact location in the DNA strand and at a precise moment in time – explains Ylli Doksani, first author of the study – We can then directly study the DNA repair machinery that works to repair this damage. Using this approach we have been able to examine the repair mechanism during chromosomal replication, the most delicate phase of the cell cycle”.
The team of IFOM scientists focused their research on isolated DNA breakpoints in order to understand better the mechanisms underlying genomic instability and the development of cancer. The study holds promising prospects for cancer prevention and therapy: “On the cancer prevention front, the first step – declares Foiani – will be to develop methods for preventive diagnosis. The identification of the set of genes involved in DNA repair mechanisms will allow us to develop ad hoc genetic tests for individuals with a family history of the AT syndrome or similar, less well known, diseases, such as ATLD. Instead, on the cancer therapy front – continues Foiani – the goal is to develop personalized therapies. The administration of common chemotherapies to individuals heterozygous for AT, for example, could have the opposite desired effect: classical chemotherapies induce stress at the level of chromosomes, which often leads to breaks in DNA. In AT individuals this can be damaging due to the lack of active DNA repair proteins. Therefore, new therapeutic targets need to be identified in order to develop more specific anti-cancer therapies in the furture.”
Original publication: Ylli Doksani, Rodrigo Bermejo, Simona Fiorani, James E. Haber e Marco Foiani; "Replicon Dynamics, Dormant Origin Firing, and Terminal Fork Integrity after Double Strand Break Formation"; Cell 2009.

Other news from the department science





















Most read news










More news from our other portals






















Last viewed contents

A promising target for new RNA therapeutics now accessible - Chemists have identified the first inhibitors of the cancer-related RNA-modifier METTL16 and have thus taken a first step towards new therapeutic options
Watermelon genome decoded - Decoded genome paves way for better watermelons
Researchers shed light on pathway from virus to brain disease
Epigenomics Septin9 Biomarker detects Colon Cancer equally in both sides of the Colon
Lorus Therapeutics allowed European patent to protect unique tumor suppressor

A 3-D printed shape-shifting smart gel
Tooth fillings of the future may incorporate bioactive glass
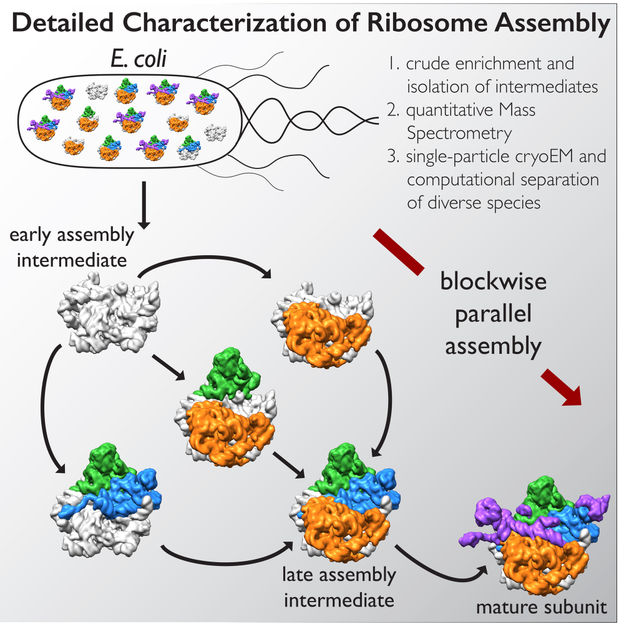
Multi-institutional collaboration uncovers how molecular machines assemble