Algorithms Offer Insight into Cellular Development
Advertisement



Neuherberg, August 31, 2016. Through RNA sequencing, researchers can measure which genes are expressed in each individual cell of a sample. A new statistical method allows researchers to infer different developmental processes from a cell mixture consisting of asynchronous stages. This finding has been published by researchers of Helmholtz Zentrum München in collaboration with colleagues from Technical University Munich.
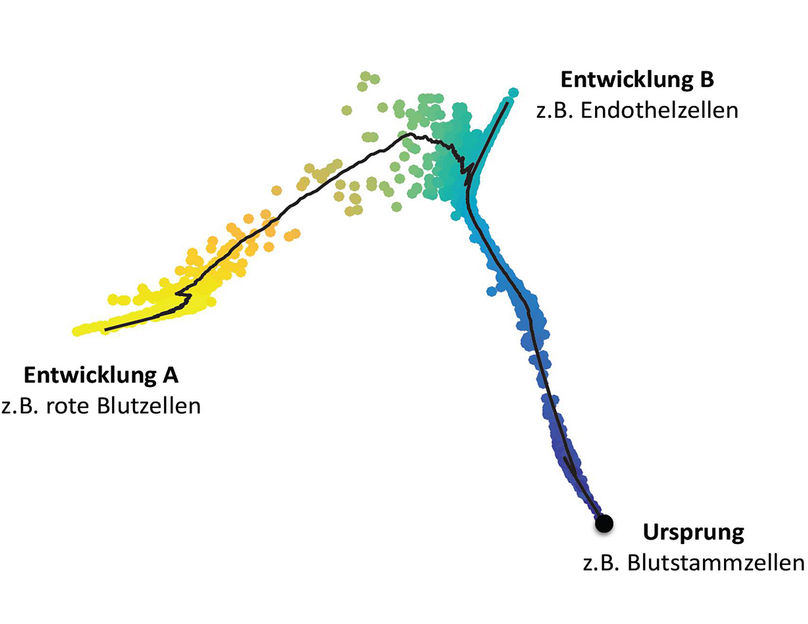
Development of cluster of blood stem cells into different cell types.
HMGU
Today, cell biology no longer focuses only on static states, but rather seeks to understand the dynamic development of cells. One example for this is the formation of various types of blood cells, such as red blood cells or endothelial cells from their precursors, the blood stem cells. To understand how this process is genetically controlled, scientists analyze which genes are expressed by means of transcriptome analysis.
“To me, it’s still amazing that we are now even able to determine the transcriptome of single cells,” said lead author Laleh Haghverdi, “especially when one realizes that a typical cell contains only a few picograms of RNA.” The availability of these data is now beginning to revolutionize many fields of research, but new statistical methods are required to interpret these correctly. “For example, all cells of a sample never start their development synchronously, and their development takes different lengths of time. Therefore, we are always dealing with a dynamic mixture,” added Haghverdi, doctoral student at the Institute of Computational Biology (ICB) at Helmholtz Zentrum München. “It is immensely difficult to construct multiple steps of a process from this, especially since the cells are only available for one measurement.”
Welcome to the era of pseudotime
To decrypt developmental processes from the measurement of a single time point, quasi a snapshot measurement, the researchers led by ICB Director Prof. Dr. Dr. Fabian Theis developed an algorithm called diffusion pseudotime to interpret single cell sequencing data. This algorithm orders cells on a virtual timeline – the pseudotime – along which they show continuous changes in the transcriptome. Thus, it can be reconstructed which genes are expressed sequentially. By means of this method, researchers can graphically display the branching lineages of the developmental paths of different cell types.
“For example, we can show how a relatively uniform cluster of blood stem cells develops into different cell types,” said study leader Theis. “While some become red blood cells, others differentiate into endothelial cells. We can trace these fates based on the transcriptome data of the single cells.” In addition, the scientists obtain information about which gene switches underlie the developments. The relatively diffuse mixture of cells which were found to be at different stages of their development can be disentangled on the computer and, after the analysis, provides a clear picture of the ongoing individual steps.
However, this is only the beginning for the researchers because the processes of blood formation are relatively well understood. They served only as a test object to determine how well the method works. “In the future we want to focus on processes that have remained elusive until now or which may not have been discovered at all,” said Theis.
Original publication




Other news from the department science




















Most read news












More news from our other portals




















Last viewed contents
Parkinson's_disease
How Do Tanning Lotions Work?
InfraReDx, Inc. - Burlington, USA

Microplastics in freshwaters - The risks to the environment are (still) low
Progress toward Artificial Photosynthesis? - Direct activation with graphitic carbon nitride makes carbon dioxide accessible for chemical synthesis
History_of_the_horse_in_South_Asia
Brain modulyzer provides interactive window into the brain
Completely Biological, yet Artificial - A new class of synthetic biomaterials made of sugar and peptide building blocks
Total_fertility_rates_by_federal_subjects_of_Russia

Not all myocarditis is the same
