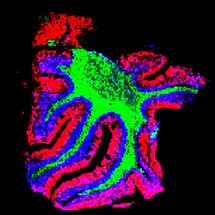
'Chromosomal Chaos:' Complex array of mutations found in rare, aggressive leukemia
Sezary syndrome (SS), an aggressive leukemia of mature T cells, is more complicated at a molecular level than ever suspected, according to investigators from the Perelman School of Medicine at the University of Pennsylvania. With a poor prognosis and limited options for targeted therapies, fighting SS needs new treatment approaches. The team's results uncover a previously unknown, complex genomic landscape of this cancer, which can be used to design new personalized drug regimens for SS patients based on their unique genetic makeup.
Sezary syndrome is a rare condition: Its incidence is estimated to be about 0.3-2 cases per 100,000 in the United States each year, and those patients have a five-year survival rate of less than 30 percent. Penn Medicine has the one of the largest referral clinics for treatment of SS patients in the country.
Taking a thorough approach to find SS mutations, senior authors Megan S. Lim, MD, PhD, a professor of Pathology and Laboratory Medicine, and Kojo Elenitoba-Johnson, MD, the Peter C. Nowell, M.D. Professor and director of the Center for Personalized Diagnostics, were not disappointed. "We basically found chromosomal chaos in all of our samples," Elenitoba-Johnson said.
The team integrated three, complementary gene sequencing approaches to look for mutations in tumor cells from SS patients: whole-genome sequencing in six subjects, sequencing of all protein-coding regions (exomes) in 66 subjects, and comparing variation in the number of copies of all genes across the genome in 80 subjects.
"We did not expect the degree of genetic complexity that we found in our study," Elenitoba-Johnson added. They identified previously unknown recurrent loss-of-function mutations that target genes regulating epigenetic pathways - ones that act on how tightly or loosely chromosomes are wound and thus accessible for genes to be expressed. One of these targets is called ARID1A, and they found that loss-of-function mutations and/or deletions in ARID1A occurred in over 40 percent of the SS genome studied.
They also identified "gain-of-function" mutations in PLCG1, and JAK1, JAK3, STAT3 and STAT5B. In preliminary drug-mutation matching studies, they found that JAK1-mutated SS cells were sensitive to JAK inhibitors, drugs that are currently approved for treatment of other hematologic cancers such as polycythemia vera and myelofibrosis.
"With knowledge like this, we can design clinical trials using JAK inhibitors for SS patients based on their JAK mutations," said Elenitoba-Johnson. "But this is just the start. These results highlight the genetic vulnerabilities that we can use in designing precision medicine therapies."
The Penn team, in collaboration with Alain Rook, MD, director of the Cutaneous T-cell Lymphoma Program and a professor of Dermatology, aims to develop a molecular taxonomy for mutations in SS patients. With the state-of-the-art DNA sequencing technology used in this study, they will be able to pinpoint the exact mistake in each patient's SS-related genes. From this, they will also be able to identify distinct subsets of the disease to stratify patients for precision therapy based on their unique mutations and the inhibitors available for those mutations.
Original publication

Other news from the department science