Drugs behave as predicted in computer model of key protein, enabling cancer drug discovery
Advertisement



Drugs important in the battle against cancer responded the way they do in real life and behaved according to predictions when tested in a computer-generated model of one of the cell's key molecular pumps -- the protein P-glycoprotein, or P-gp.
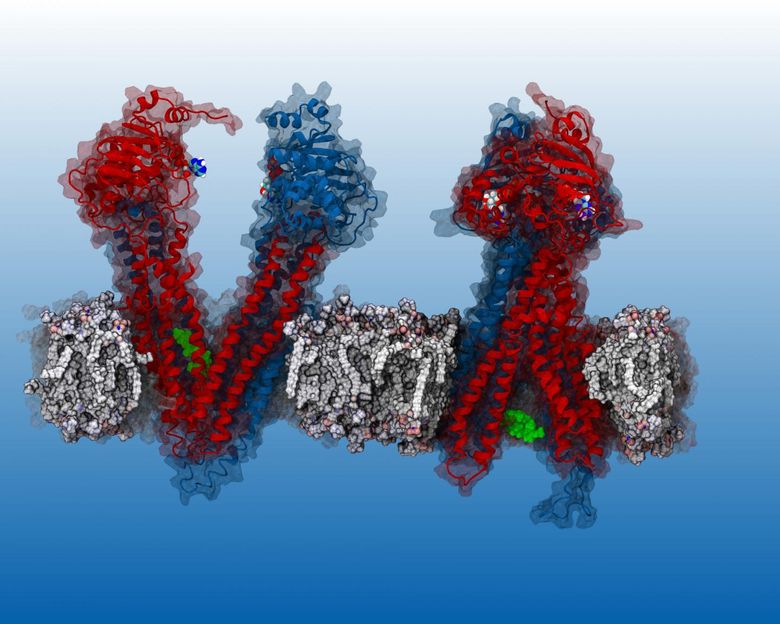
Verapamil (green blob), inhibits the P-gp pump. Until now, the workings of the pump could not be observed so researchers didn't know exactly where Varapamil 'binds' in P-gp. SMU researchers report that their simulation reveals the binding sites.
James McCormick
Biologists at Southern Methodist University, Dallas, developed the computer generated model to overcome the problem of relying on only static images for the structure of P-gp, said biologist John G. Wise, lead author on the journal article announcing the advancement.
The new SMU model allows researchers to dock nearly any drug in the P-gp protein and see how it will actually behave in P-gp's pump, said Wise, an associate professor in SMU's Department of Biological Sciences.
"The value of this fundamental research is that it generates dynamic mechanisms that let us understand something in biochemistry, in biology," he said. "And by understanding P-gp in such detail, we can now think of ways to better and more specifically inhibit it."
P-gp is the cellular pump that protects cells by pumping out toxins. But that's a problem when P-gp targets chemotherapy drugs as toxic, preventing chemo from killing cancer cells. Scientists are searching for ways to inhibit P-gp's pumping action.
The SMU researchers tested Tariquidar, a new P-gp inhibitor still in clinical trials. Inhibitors offer hope for stopping P-gp's rejection of chemotherapeutics by stalling the protein's pumping action. Pharmacology researchers disagree, however, on where exactly Tariquidar binds in P-gp.
When run through the SMU model, Tariquidar behaved as expected: It wasn't effectively pumped from the cell and the researchers observed that it prefers to bind high in the protein.
"Now we have more details on how Tariquidar inhibits P-gp, where it inhibits and what it's actually binding to," Wise said.
Also using the model, the researchers discovered greater detail than previously known about the behavior of other drugs as well, and how those drugs bind in P-gp to stop its pumping action.
Testing the virtual P-gp model by virtually docking real drugs Wise and his colleagues tested one of the workhorse drugs of chemotherapy, daunorubicin, a close cousin of Adriamycin. An aggressive chemotherapeutic, daunorubicin stops DNA replication in the cell, and is a classic target for P-gp to pump out of a cell, Wise said.
"For a long time, it's been thought that there are at least a couple of distinct binding sites for drugs," Wise said. "Sure enough, with our models, we found that daunorubicin, at least, prefers to bind on one side of the P-gp model, while verapamil - a commonly prescribed blood pressure medicine - prefers the other side."
Not only did the researchers show computationally that there are two different starting points for drugs, they also showed that there are two different pathways to get the drugs through.
"The two different drugs start at different sites and they're funneled to the outside by being pushed by the protein," Wise said. "But the actual parts of the protein that are pushing the drugs out are different."
Original publication

Other news from the department science



















































