First, do no harm: Study finds danger in standard treatment for a serious lung disease
Results show importance of rigorous, placebo-controlled, independent evaluation of treatments for any disease
Advertisement



A combination of three drugs used worldwide as the standard of care for a serious lung disease puts patients in danger of death or hospitalization, and should not be used together to treat the disease, called idiopathic pulmonary fibrosis, according to the surprising results of a rigorous independent study. The study in the New England Journal of Medicine was conducted by IPF Clinical Research Network, funded by the National Heart, Lung, and Blood Institute of the National Institutes of Health.
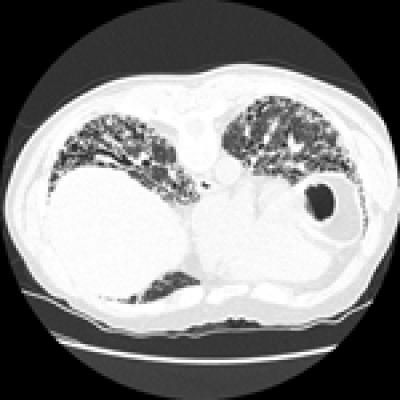
This CT scan shows the "honeycomb" pattern of scarring in the lungs of a patient with idiopathic pulmonary fibrosis, a progressive disease with few good treatment options. A new study has found that a three-drug combination used worldwide to treat IPF is in fact associated with a high risk of death and hospitalization than placebo.
University of Michigan Health System
"The findings show the importance of testing even those treatments that doctors give routinely for any type of condition - to see if they truly help, and don't harm, patients," says University of Michigan Health System lung specialist Fernando Martinez, M.D., who will present the results.
Martinez and his colleagues report that patients in the mild to moderate stages of the progressive lung-scarring disease had a far higher chance of dying or being hospitalized if they were taking a three-drug combination used worldwide, compared with those taking a placebo.

What's more, the three-drug combo yielded no improvement in lung function, or even slowing of loss of lung function, compared with placebo. Results from a group taking the single drug, N-acetylcysteine (NAC), are still being gathered and analyzed.
This evidence is from a randomized, placebo-controlled, double-blind, federally funded trial that included patients with a definitive diagnosis of IPF who were treated at 25 centers taking part in the IPF Clinical Research Network or IPFNet. The study was stopped early when an interim analysis showed signs of harm from the three-drug combination of prednisone, azathioprine and NAC.
The findings should cause physicians worldwide to stop using this combination to treat IPF patients similar to those in the trial, say the authors.
And, the dramatic finding of harm from a standard treatment should cause physicians to apply rigorous testing methods to other types of treatment, and highlights the importance of independent federal funding for such studies, says Martinez. The authors salute the volunteer IPF patients who agreed to be randomly assigned to a treatment or placebo for 60 weeks.
Martinez, an internationally known IPF researcher and clinician in the U-M Medical School's Division of Pulmonary Medicine, remarks that results will soon be known for the group taking NAC alone, compared with those taking placebo. The current paper and presentation do not include results from this group.
In the results presented this week, the authors report that eight patients in the group of 77 assigned to the three-drug combination died, compared with one in the placebo group. A total of 23 of the three-drug patients were hospitalized during the trial, compared with 7 in the placebo group. There was no sign that the three-drug combination slowed the progression of IPF or improved lung function, as measured by forced vital capacity.
The study is called PANTHER-IPF, for Prednisone, Azathioprine, and N-Acetylcysteine: a Study That Evaluates Response in Idiopathic Pulmonary Fibrosis. Except for a donation of NAC and a matched placebo by the company that makes the drug, there was no industry support for the work.
IPF, which affects nearly 100,000 Americans, slowly steals the ability to breathe freely. Its cause or causes are not clear, which is why it is called "idiopathic." Over time it leads to the buildup of scar tissue in the lungs that accumulates in a distinctive honeycomb pattern that can be seen on biopsy or CT scan. It is known as an interstitial lung disease because it affects the tissue around the air sacs in the lungs.
IPF patients live an average of five years after diagnosis, though a lung transplant at a center such as U-M's Transplant Center can extend life for years beyond. Most patients are over the age of 65 when diagnosed, but IPF can strike younger people as well.
Because lung transplants are such a dramatic and rarely available therapy, researchers at U-M and other centers are working to find new treatments while also studying the underlying biological factors in the disease. The PANTHER-IPF trial was designed to test a standard therapy in a rigorous way.




Other news from the department science