Protein droplets may cause many types of genetic disease
Malfunction of cellular condensates is a disease mechanism relevant for congenital malformations, common diseases, and cancer
Most proteins localize to distinct protein-rich droplets in cells, also known as “cellular condensates”. Such proteins contain sequence features that function as address labels, telling the protein which condensate to move into. When the labels get screwed up, proteins may end up in the wrong condensate. According to an international team of researchers from clinical medicine and basic biology, this could be the cause of many unresolved diseases. The findings appeared in the journal Nature.
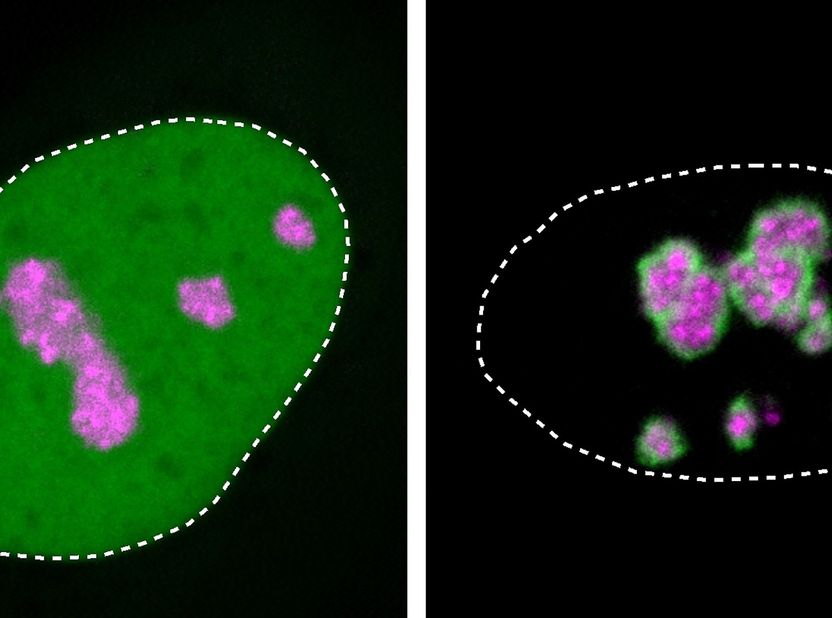
Close-ups of cell nuclei in a human cell culture: HMGB1 protein (green) is usually found throughout the nucleus (dotted line). Mutant HMGB1, shown on the right, preferentially localizes at the nucleolus (marked in magenta) and forms a solidified layer over it, which causes disease.
Henri Niskanen, MPIMG
Patients with BPTA syndrome have characteristically malformed limbs featuring short fingers and additional toes, missing tibia bones in their legs and reduced brain size. As the researchers found out, BPTAS is caused by a special genetic change that causes an essential protein to migrate to the nucleolus, a large proteinaceous droplet in the cell nucleus. As a result, the function of the nucleolar condensate is inhibited and developmental disease develops.
“What we discovered in this one disease might apply to many more disorders. It is likely not a rare unicorn that exists only once. We just could not see the phenomenon until now because we did not know how to look for it,” says Denise Horn, a clinical geneticist at the Institute of Medical and Human Genetics at Charité – Universitätsmedizin Berlin.
In collaboration with scientists at the Max Planck Institute for Molecular Genetics (MPIMG) in Berlin, the University Hospital Schleswig-Holstein (UKSH), and contributors from all around the world, the team is pushing open a door to new diagnoses that could lead to the elucidation of numerous other diseases as well as possible future therapies.
“We discovered a new mechanism that could be at play in a wide range of diseases, including hereditary diseases and cancer,” says Denes Hnisz, Research Group Leader at the MPIMG. “In fact, we have discovered over 600 similar mutations, 101 of which are known to be associated with different disorders.”
“The actual work is just starting now,” adds human geneticist Malte Spielmann of UKSH in Lübeck and Kiel. “We will find many more genes with such disease-causing mutations and can now test their mode of action.”
An unusual mutation
Affected individuals have complex and striking malformations of the limbs, face, and nervous and bone systems, only partially described by the already-long disease name “brachyphalangy-polydactyly-tibial aplasia/hypoplasia syndrome” (BPTAS).
“With fewer than ten documented cases worldwide, the disease is not only rare, but ultra-rare,” says Martin Mensah, clinical geneticist at the Institute of Medical and Human Genetics at Charité. To track down the cause, he and his colleagues decoded the genome of five affected individuals and found that the gene for the protein HMGB1 was altered in all patients.
This protein has the task of organizing the genetic material in the cell nucleus and facilitates the interaction of other molecules with the DNA, for example to read genes.
In mice, a complete loss of the gene on both chromosomes is catastrophic and leads to death of the embryo. In some patients with only one copy mutated, however, the cells are using the intact copy on the other chromosome, resulting only in mild neurodevelopmental delay. But the newly discovered cases did not fit this scheme.
“All five unrelated individuals featured the same ultra-rare disorder and had virtually the same mutation”, says Mensah, who is a fellow of the Clinician Scientist Program operated by the Berlin Institute of Health at Charité (BIH) and Charité. “This is why we are sure that the HMGB1 mutation is the cause of the disease. However, at that point, we had no clue how the gene product functionally caused disease, especially given that loss-of-function mutations were reported to result in other phenotypes.”
Charged protein extensions
A closer look revealed that different mutations of HMGB1 have different consequences. The sequencing data showed that in the affected individuals with the severe malformations, the reading frame for the final third of the HMGB1 gene is shifted.
After translation to protein, the corresponding region is now no longer equipped with negative but with positively charged amino acid building blocks. This can happen if a number of genetic letters not divisible by three is missing in the sequence, because exactly three consecutive letters always code for one building block of the protein.
However, the tail part of the protein does not have a defined structure. Instead, this section hangs out of the molecule like a loose rubber band. The purposes of such protein tails (also called “intrinsically disordered regions”) are difficult to study because they often become effective only in conjunction with other molecules. So how might their mutation lead to the observed disease?
Protein droplets in the cell
To answer this question, the medical researchers approached biochemists Denes Hnisz and Henri Niskanen at the MPIMG, who work with cellular condensates that control important genes. These droplet-like structures behave much like the oil and vinegar droplets in a salad dressing. Composed of a large number of different molecules, they are separated from their surroundings and can undergo dynamic changes.
“We think condensates are formed in the cell for practical reasons,” Niskanen explains. Molecules for a specific task are grouped together in this way, say to read a gene. For this task alone, he says, several hundred proteins need to somehow make their way to the right place.
“Intrinsically disordered regions, which tend not to have an obvious biochemical role, are thought to be responsible for forming condensates,” Niskanen says, giving an example to describe how important the physical properties of the protein extensions are in this regard. “I can easily make a ball from many loose rubber bands that holds together relatively tightly and that can be taken apart with little effort. A ball of smooth fishing line or sticky tape, on the other hand, would behave quite differently.”
Solidifying droplets
The nucleolus within the cell nucleus is also a condensate, which appears as a diffuse dark speck under the microscope. This is where many proteins with positively charged tails like to linger. Many of these provide the machinery required for protein synthesis, making this condensate essential for cellular functions.
The mutant protein HMGB1 with its positively charged molecular tail is attracted to the nucleolus as well, as the team observed from experiments with isolated protein and with cell cultures.
But since the mutated protein region has also gained an oily, sticky part, it tends to clump. The nucleolus loses its fluid-like properties and increasingly solidifies, which Niskanen was able to observe under the microscope. This impaired the vital functions of the cells – with the mutated protein, more cells in a culture died compared to a culture of cells without the mutation.
Combing through databases
The research team then searched databases of genomic data from thousands of individuals looking for similar incidents. In fact, the scientists were able to identify more than six hundred similar mutations in 66 proteins, in which the reading frame had been shifted by a mutation in the protein tail, making it both more positively charged and more “greasy”. Of the mutations, 101 had previously been linked to several different disorders.
For a cell culture assay, the team selected 13 mutant genes. In 12 out of 13 cases, the mutant proteins had a preference to localize into the nucleolus. About half of the tested proteins impaired the function of the nucleolus, resembling the disease mechanism of BPTA syndrome.
New explanations for existing diseases
“For clinical research, our study could have an eye-opening effect,” says Malte Spielmann, who led the research together with Denes Hnisz and Denise Horn. “In the future, we can certainly elucidate the causes of some genetic diseases and hopefully one day treat them.”
However, “congenital genetic diseases such as BPTAS are almost impossible to cure even with our new knowledge”, says Horn. “Because the malformations already develop in the womb, they would have to be treated with drugs before they develop. This would be very difficult to do.”
But tumor diseases are also predominantly genetically determined, adds Hnisz: “Cellular condensates and the associated phase separation are a fundamental mechanism of the cell that also plays a role in cancer. The chances of developing targeted therapies for this are much better.”
Original publication
Other news from the department science





















































