Abundant ‘secret doors’ on human proteins could reshape drug discovery
Identification of hidden vulnerabilities on surface of ‘undruggable’ proteins could transform treatment of disease
The number of potential therapeutic targets on the surfaces of human proteins is much greater than previously thought, according to the findings of a new study in the journal Nature.
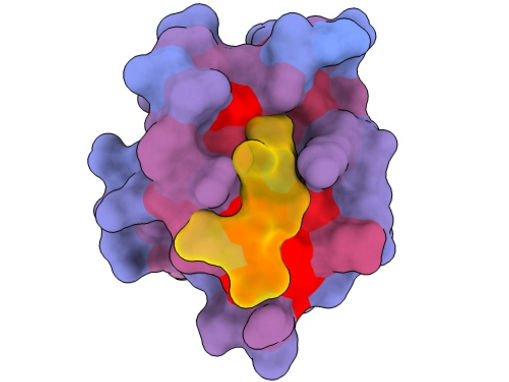
A three-dimensional image showing the human protein PSD95-PDZ3 from different angles. Shown is the binding partner CRIPT (yellow) in the active site with the blue-to-red colour gradient indicating increasing potential for allosteric effects. Based on PDB accession 1BE9.
André Faure/CRG
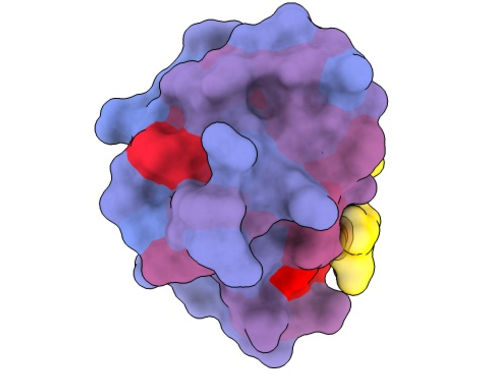
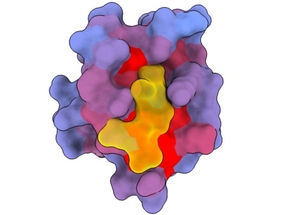
A three-dimensional image showing the human protein PSD95-PDZ3 from different angles. Shown is the binding partner CRIPT (yellow) in the active site with the blue-to-red colour gradient indicating increasing potential for allosteric effects. Based on PDB accession 1BE9.
André Faure/CRG
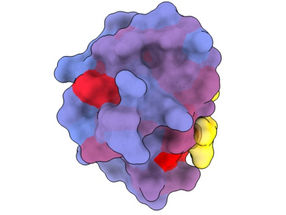
A ground-breaking new technique developed by researchers at the Centre for Genomic Regulation (CRG) in Barcelona has revealed the existence of a multitude of previously secret doors that control protein function and which could, in theory, be targeted to dramatically change the course of conditions as varied as dementia, cancer and infectious diseases.
The method, in which tens of thousands of experiments are performed at the same time, has been used to chart the first ever map of these elusive targets, also known as allosteric sites, in two of the most common human proteins, revealing they are abundant and identifiable.
The approach could be a game changer for drug discovery, leading to safer, smarter and more effective medicines. It enables research labs around the world to find and exploit vulnerabilities in any protein – including those previously thought ‘undruggable’.
“Not only are these potential therapeutic sites abundant, there is evidence they can be manipulated in many different ways. Rather than simply switching them on or off, we could modulate their activity like a thermostat. From an engineering perspective, that’s striking gold because it gives us plenty of space to design ‘smart drugs’ that target the bad and spare the good,” explains André Faure, postdoctoral researcher at the CRG and co-first author of the paper.
Proteins play a central role in all living organisms and carry out vital functions such as providing structure, speeding up reactions, acting as messengers or fighting disease. They are made of amino acids, folding into countless different shapes in three-dimensional space. The shape of a protein is crucial for its function, with just one mistake in an amino acid sequence resulting in potentially devastating consequences for human health.
Allostery is one of the great unsolved mysteries of protein function. Allosteric effects occur when a molecule binds to the surface of a protein, which in turn causes changes at a distant site in the same protein, regulating its function by remote control. Many disease-causing mutations, including numerous cancer drivers, are pathological because of their allosteric effects.
Despite their fundamental importance, allosteric sites are incredibly difficult to find. This is because the rules governing how proteins work at the atomic level are hidden out of sight. For example, a protein might shapeshift in the presence of an incoming molecule, revealing hidden pockets deep within its surface that are potentially allosteric but not identifiable using conventional structure determination alone.
Drug hunters have traditionally designed treatments that target a protein’s active site, the small region where chemical reactions occur or targets are bound. The downside of these drugs, also known as orthosteric drugs, is that active sites of many proteins look very similar and so drugs tend to bind and inhibit many different proteins at once, leading to potential side effects. In comparison, the specificity of allosteric sites means that allosteric drugs are some of the most effective types of medication currently available. Many allosteric drugs, which treat various conditions ranging from cancer to AIDS to hormone disorders, have been discovered by accident.
The authors of the study addressed this challenge by developing a technique called double deep PCA (ddPCA), which they describe as a ‘brute force experiment’. “We purposefully break things in thousands of different ways to build a complete picture of how something works”, explains ICREA Research Professor Ben Lehner, Coordinator of the Systems Biology programme at the CRG and author of the study. “It’s like suspecting a faulty spark plug, but instead of only checking that, the mechanic dismantles the entire car and checks it piece by piece. By testing ten thousand things in one go we identify all the pieces that really matter.”
The method works by changing the amino acids that make up a protein, resulting in thousands of different versions of the protein with just one or two differences in the sequence. The effects of the mutations are then tested all at the same time in living cells in the laboratory.
“Each cell is a tiny factory making a different version of the protein. In a single test tube we have millions of different factories and so we can very rapidly test how well all the different versions of a protein work”, adds Dr. Lehner. The data collected from the experiments is fed into neural networks, algorithms that analyze data by mimicking the way the human brain operates, which result in comprehensive maps that pinpoint the location of allosteric sites on the surfaces of proteins.
One of the great advantages of the method is that it is an affordable technique accessible to any research lab around the world. “It massively simplifies the process needed to find allosteric sites, with the technique working at a level of accuracy better than several different more expensive and time-consuming lab methods”, says Júlia Domingo, co-first author of the study. “Our hope is that other scientists use the technique to rapidly and comprehensively map the allosteric sites of human proteins one by one.”
One of the longer-term benefits of the technique is its potential to study the function and evolution of proteins. The authors of the study believe that, if scaled up, the method could one day result in advances that can precisely predict the properties of proteins from their amino acid sequences. If successful, the authors argue this would usher in a new era of predictive molecular biology, allowing much faster development of new medicine and clean, biology-based industry.
“While some tools can predict a protein’s structure by reading its sequence, our method goes one step further by telling us how a protein works. This is part of a bigger vision to make biology as engineerable as aeroplanes, bridges or computers. We have faced the same challenges for over 70 years, but it turns out they are more tractable than we previously thought. If we succeed it will open a new field with unprecedented possibilities”, concludes Dr. Lehner.
Original publication
Other news from the department science