Inhibiting tumour-specific repair mechanisms
Findings can help to develop new or to improve existing strategies in cancer therapy
Advertisement


Mutations of the BRCA1 and BRCA2 genes – also known as “breast cancer genes” – are associated with the development of inherited breast cancer as well as other types of tumours. A group of researchers from the Technical University of Darmstadt, the University of California and the University of Texas, characterized the specific repair processes in BRCA2-mutated tumour cells providing findings that can help to develop new or to improve existing strategies in cancer therapy. The results of this study were recently published in the renowned science journal Nature Cell Biology (NCB).
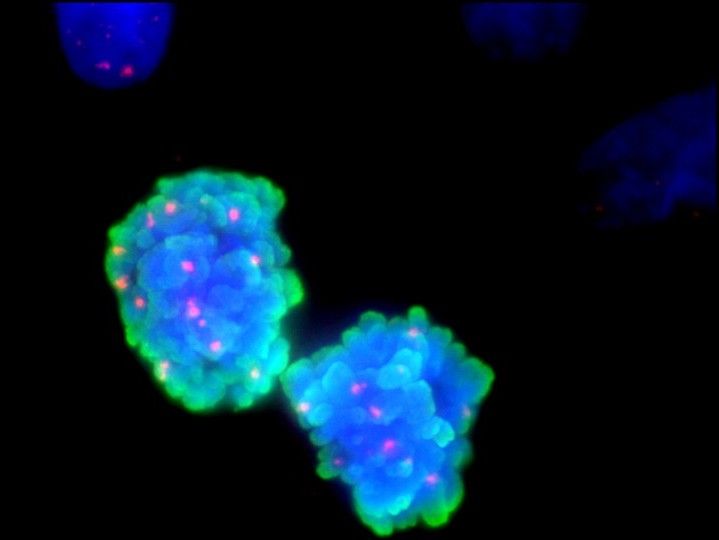
Double-strand breaks during mitosis. Two dividing BRCA2-mutated cells are shown in which the TMEJ repair process was inhibited. The cells exhibit many unrepaired double-strand breaks (shown by the red dots), that are hardly detected in cells with functional BRCA2 and that can cause cell death. The DNA was stained in blue, the mitosis marker pH3 was stained in green und the double-strand break marker yH2AX was stained in red.
Michael Ensminger
The DNA which encodes the genetic information of each cell is constantly damaged by endogenous and exogenous influences. The thousands of DNA lesions arising every day in each cell of our body are effectively detected and repaired by different cellular mechanisms called the DNA damage response. Defects of these mechanisms can be associated with cancer development.
The most severe DNA lesion is the DNA double-strand break as the DNA molecule is completely raptured. One double-strand break can already be sufficient to kill a cell if it stays unrepaired or if it is repaired in an error-prone manner. Homologous recombination is one of two main repair pathways for repairing double-strand breaks and BRCA1 and BRCA2 genes are essential for this process. Thus, this repair pathway is defective in tumour cells harbouring mutations for BRCA1 or BRCA2.
Targeted induction of double-strand breaks in cancer cells
The aim of a cancer therapy is to efficiently kill tumour cells by inducing double-strand breaks using ionizing radiation or chemotherapeutics. BRCA2-mutated tumour cells cannot repair these double-strand breaks via homologues recombination and strongly rely on alternative repair pathways. A promising new strategy in cancer therapy could be the specific inactivation of these alternative repair processes. This would selectively kill the BRCA2-mutated tumour cells and not the healthy cells in our bodies, as BRCA2 is not altered in these cells and therefore they can still repair double-strand breaks via homologous recombination. However, the alternative repair processes in BRCA2-mutated cells are not fully understood preventing a specific inactivation.
New insights into repair processes
The research group of Professor Markus Löbrich at TU Darmstadt has characterized a specific alternative repair process in BRCA2-mutated cells. This study was recently published in the renowned science journal Nature Cell Biology. Together with research groups of the University of California and the University of Texas, Löbrich and colleagues demonstrate that a repair process called POLθ-mediated end-joining (TMEJ) is responsible for repairing double-strand breaks in BRCA2-mutated cells.
If this process is inhibited, double-strand breaks in BRCA2-mutated cells stay unrepaired subsequently causing cell death. Moreover, two factors were identified which regulate the activation of TMEJ: RAD52 and BRCA2 itself. If RAD52 is inhibited in BRCA2-mutated cells, TMEJ in performed in an error-prone manner causing fusions between different chromosomes which represent potentially lethal aberrations contributing to cell death.
Original publication

Other news from the department science























































