Unlocking the secrets of plant genomes in high resolution
New software tool allows for precise assignment to the correct copies – a process known as ‘phasing’
Advertisement



Resolving genomes, particularly plant genomes, is a very complex and error-prone task. This is because there are several copies of all of the chromosomes and they are very alike. A team of bioinformatics researchers from Heinrich Heine University Düsseldorf (HHU) has now developed a software tool that allows for precise assignment to the correct copies – a process known as ‘phasing’.

The new software tool makes it possible to determine the genome of species such as potatoes with a high degree of accuracy (symbolic image).
Capri23auto, pixabay.com

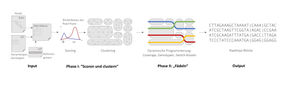
How the bioinformatics software tool ‘WhatsHap polyphase’ works.
Gunnar Klau/HHU

The genomes of all higher life forms are stored in the cell nucleus on chromosomes. Chromosomes are composed of strands of the DNA molecule. The genetic information itself is encoded in a sequence of adjacent base pairs of the molecules adenine (A), cytosine (C), guanine (G) and thymine (T).
Different species have different numbers of chromosomes; for example, humans have 23, while potatoes have 12 and wheat has 7. In addition, there are different copies or ‘haplotypes’ of the chromosomes. Humans have two copies, one from the mother and one from the father, while potatoes have four and wheat even has six. Species with two copies are referred to as ‘diploid’, whereas those with more than two are ‘polyploid’. The copies are almost identical, with ‘almost’ being the operative word. It is the differences between them that determine the variability of the organisms within a population.

In order to unlock the genetic information, the researchers tackled something akin to a large jigsaw: They took a larger number of cells, divided the cells’ genomes into lots of small parts – called ‘reads’ – and sequenced the information contained in those parts. This was necessary because the technology currently available can only process small sections of DNA.
The result was a huge volume of data – billions of reads, with a data volume of several hundred gigabytes. They comprise sequences of differing lengths made up of the letters A, C, G and T. The next task for the bioinformatics researchers was to determine their position within a chromosome, then assign the corresponding sections to a chromosome (a process known as ‘mapping’) and finally to find the right copies of the chromosome. This last stage is known as ‘phasing’. The task is made more difficult by sequencing errors.
There are good, efficient tools available for mapping. However, the bioinformatics tools needed for phasing are still in their infancy. This was precisely where the team of bioinformatics researchers from HHU focused their attention. In a joint project funded by the German Research Foundation and managed by Prof. Dr. Gunnar Klau (Algorithmic Bioinformatics working group) and Prof. Dr. Tobias Marschall (Institute of Medical Biometry and Bioinformatics, University Hospital Düsseldorf) in collaboration with Prof. Dr. Björn Usadel (Institute of Biological Data Science), they developed a software tool named ‘WhatsHap polyphase’ and tested the tool successfully using model data as well as the potato genome.
This new tool solves the problem using a two-phase process. The first phase involves clustering the reads, i.e. splitting them into groups. Reads in one group probably come from one haplotype or a region of identical haplotypes. The second phase involves ‘threading’ the haplotypes through the clusters. During threading, the reads are assigned to the haplotypes as evenly as possible, ensuring as little as possible jumping back and forth between clusters.
The new tool has been added to the main ‘WhatsHap’ package, which is freely available. The package has already been used to carry out the phasing successfully for diploid chromosome sets, e.g. for humans. This new addition from the team based in Düsseldorf means that phasing is now also possible for polyploid organisms. Prof. Klau said: “Our new technology allows for plant genomes to be phased in high resolution and with a low margin of error”.
Original publication




Other news from the department science




















These products might interest you