Fine-tuning an anti-cancer drug
Cancer remains a deadly threat despite the best efforts of science. New hopes were raised a few years ago with the discovery that the uncontrolled growth of cancer cells could be thwarted by blocking the action of proteasomes. Biochemists at the Technische Universitaet Muenchen (TUM) have illuminated a reaction pathway that does just that, in collaboration with researchers from Nereus Pharmaceuticals, based in San Diego, California. In the Journal of medicinal chemistry, they report insights that could potentially lead to the development of custom-tailored anti-cancer drugs.
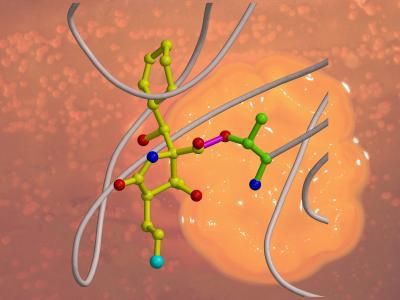
Snapshots of a biochemical process - obtained through X-ray crystallography - provide insights that could guide development of tailored anti-cancer drugs. Researchers at the Technische Universitaet Muenchen and Nereus Pharmaceuticals have determined in detail how the bacteria-derived molecule Salinosporamide A disables the intracellular waste-processing plants called proteasomes. The researchers compare the mechanism to a key that breaks off in a lock.
TUM Dept. of Biochemistry and Nereus Pharmaceuticals.
What makes cancer cells so dangerous is that they proliferate much more rapidly than other cells. An important contribution to this capability is made by a particular group of proteins, the so-called kinases. And it's against the kinases that many cancer drugs in development today take aim. Another promising approach came to light a few years ago with the discovery that the proliferation of cancer cells could also be arrested through proteasome inhibition. Yet the first drug to employ this strategy caused a number of severe side-effects.
In the search for alternatives, San Diego-based Nereus Pharmaceuticals homed in on a species of marine bacteria known as Salinispora tropica. These bacteria produce a small molecule that kills affected cells by disabling proteasomes, which serve as their waste processing plants. "In the life cycle of a cell, proteins are always being built up that will need to be demolished after they have done their work," explains TUM Professor Michael Groll, leader of the research team in Munich. "If this breakdown is blocked, the cells choke on their own waste."
After promising preclinical trials, the bacteria-produced Salinosporamide A (NPI-0052; Sal-A) has advanced into human clinical trials. "Over millions of years, the bacteria developed this substance into a perfect weapon," says Dr. Barbara Potts, vice president for chemical and oncological development at Nereus Pharmaceuticals. The ideal cancer drug would kill only cancer cells, while doing the least harm possible to healthy cells. The researchers took a closer look at the pathway for this reaction, in the hope that they might better understand the mechanism and the best approach to future generation analogues.
The research team of Barbara Potts and Michael Groll managed to produce crystals of proteasomes blocked by Salinosporamide A and determined, through X-ray crystallography, the precise arrangement of the atoms. It became clear why the bacterial poison is so effective: The molecule fits an opening in the proteasome like a key, and locks it up. A subsequent reaction transforms the molecule to a complex that can no longer be detached, in effect breaking off the key in the lock. Vital processes come to a halt.
Halogen-hydrocarbons are favored in industrial chemistry, because the halogen atom can be easily separated from other groups. It's just this trick that the Salinispora tropica bacterium employs in the case of Salinosporamide A. It uses a chloride as its so-called "leaving group" to trigger an internal reaction forming a ring-like bond. If the ring is closed, the lock is jammed.
The researchers next produced variants of Salinosporamide A and once again succeeded in crystallizing them and using X-ray techniques for structural analysis. By replacing the chlorine atom with fluorine, they were able to observe the progress of the reaction. After the key had been stuck in the lock for one hour of reaction time, the biochemists were still able to pull it out again. A few hours later, the fluorine was split off, and the lock was blocked.
"After the millions of years that have gone into the evolutionary development of this method in bacteria, it's unlikely that a better way to block the proteasome is even possible," Groll says. "Now that we know how the best possible reaction proceeds, we can alter it in targeted ways with the aim of developing tailored, effective proteasomal drugs that will have improved safety and efficacy."
Original publication: Michael Groll, Katherine A. McArthur, Venkat R. Macherla, Rama Rao Manam and Barbara C. Potts, "Snapshots of the Fluorosalinosporamide/20S Complex Offer Mechanistic Insights for Fine Tuning Proteasome Inhibition"; Journal of Medicinal Chemistry 2009.

Other news from the department science


















































