Erbgut von mehr als 1000 Bakterien bestimmt
Vergleichsdatenbank vereinfacht in Zukunft mikrobielle und metagenomische Forschung deutlich
Biologen und Informatiker des Leibniz-Instituts DSMZ−Deutsche Sammlung von Mikroorganismen und Zellkulturen und des kalifornischen Joint Genome Institute (JGI) haben in einem fünfjährigen Forschungsprojekt das komplette Erbgut von über 1000 Bakterien und Archaeen bestimmt. Nie zuvor wurden in einem einzelnen Projekt mehr mikrobielle Typstammgenome sequenziert. So wurde die Zahl der sequenzierten Typstämme, also der Referenzkulturen einzelner Bakterienarten, auf einen Schlag mehr als verdoppelt. Darüber hinaus haben die Wissenschaftler in den genetischen Codes zahlreiche neue Enzymkomplexe identifiziert, die Grundlage für neue biotechnologische oder medizinische Anwendungen sein können.
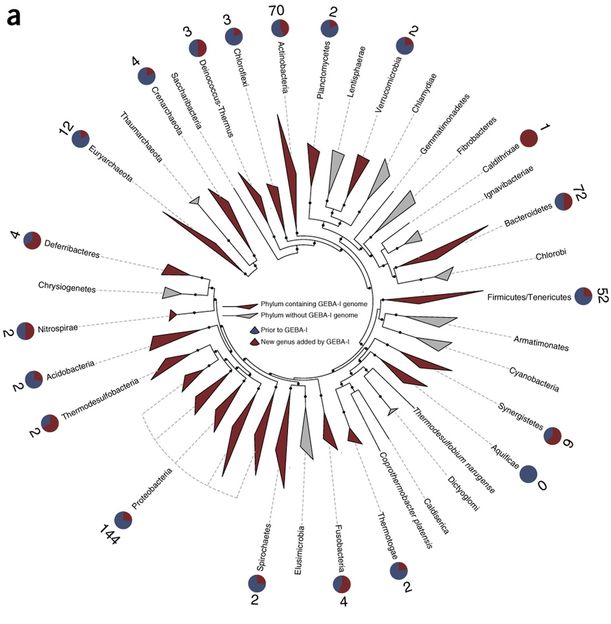
Phylogenie und Verteilung der GEBA-Stämme
Nature Biotechnology
Die Sequenzdaten stellen die Wissenschaftler im Rahmen der gemeinnützigen GEBA-Initiative (Genomic Encyclopedia of Bacteria and Archaea, Genomische Enzyklopädie der Bakterien und Archaeen) öffentlich zur Verfügung. Damit steht für Wissenschaftler weltweit eine Vergleichsdatenbank bereit, die die mikrobielle und metagenomische Forschung in Zukunft deutlich vereinfacht. Die Studie konnte auch zeigen, dass die vielen neuen Referenzgenome eine deutlich genauere Identifizierung von Sequenzen aus Umweltproben ermöglichen.
Die DSMZ brachte in das Projekt ihre Expertise in der Anzucht von Mikroorganismen und in der phylogenomischen Auswertung ein. So konnten die Experten der DSMZ auch aus schwierig zu kultivierenden Bakterienstämmen Zellmasse und DNA für die Genomsequenzierung gewinnen. Aber auch Stammbäume wurden an der DSMZ bioinformatisch aus kompletten Genomsequenzen rekonstruiert.
„Bisher lag der Fokus bei der Entschlüsselung von bakteriellem Erbgut vor allem auf einzelnen medizinisch und biotechnologisch besonders wichtigen Arten“, erläutert PD Dr. Markus Göker, Projektleiter an der DSMZ. So stammten 2015 ganze 43 Prozent aller sequenzierter Bakteriengenome von gerade einmal zehn verschiedenen Krankheitserregern. Diese Fokussierung hat zwar geholfen, ein besseres Verständnis von der Entstehung von Krankheiten zu bekommen, hat aber auch zu einem verzerrten Bild der bakteriellen Diversität geführt, insbesondere mit Blick auf ihre funktionellen Eigenschaften. Denn je weniger eng zwei Bakterienstämme miteinander verwandt sind, desto größer ist oft die Wahrscheinlichkeit, dass sie unterschiedliche physiologische Eigenschaften oder Funktionen aufweisen, so Göker. Mit den jetzt präsentierten Daten haben die Projektpartner einen ersten großen Schritt vorwärts unternommen, um die verfügbaren Daten zu bakteriellem Erbgut auf eine phylogenetisch breitere Grundlage zu stellen.
Dennoch weist der bakterielle Stammbaum noch immer große Lücken auf. DSMZ und JGI haben daher bereits drei Folgeprojekte gestartet, in denen die Partner jeweils weitere 1000 bakterielle Genome sequenzieren werden.
Originalveröffentlichung
Weitere News aus dem Ressort Wissenschaft